
Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
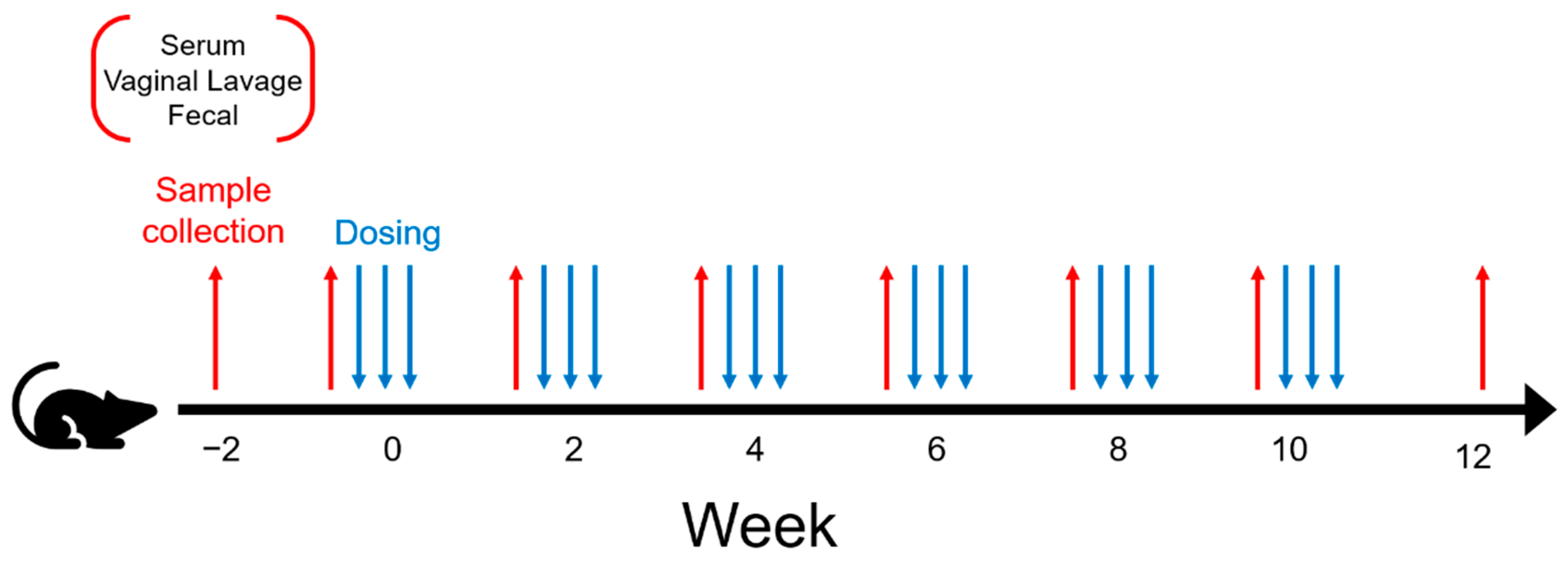
2.2. Experimental Design
2.3. Bacterial Strains and Culture Conditions
2.4. Mouse Immunization and Housing
2.5. Sample Collection and Processing
2.6. ELISA Assays
2.7. Statistical Methods for ELISA Data
2.8. IgA-Bound Bacterial Sorting
2.9. Microbiome Library Preparation
2.10. Data Processing
2.11. Diversity and Correlation Analysis
2.12. Random Forest
3. Results
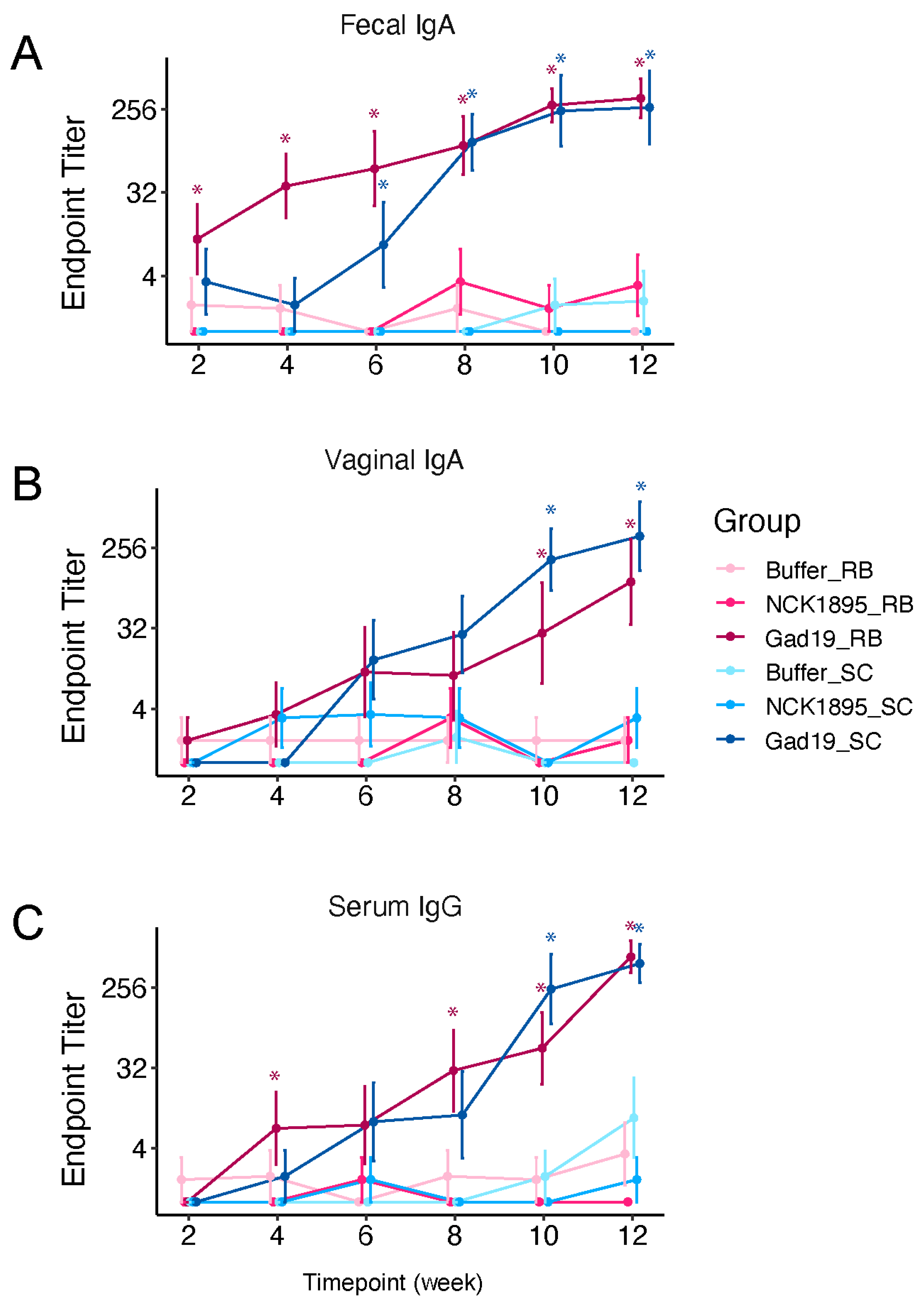
3.1. Antigen-Specific Antibody Responses
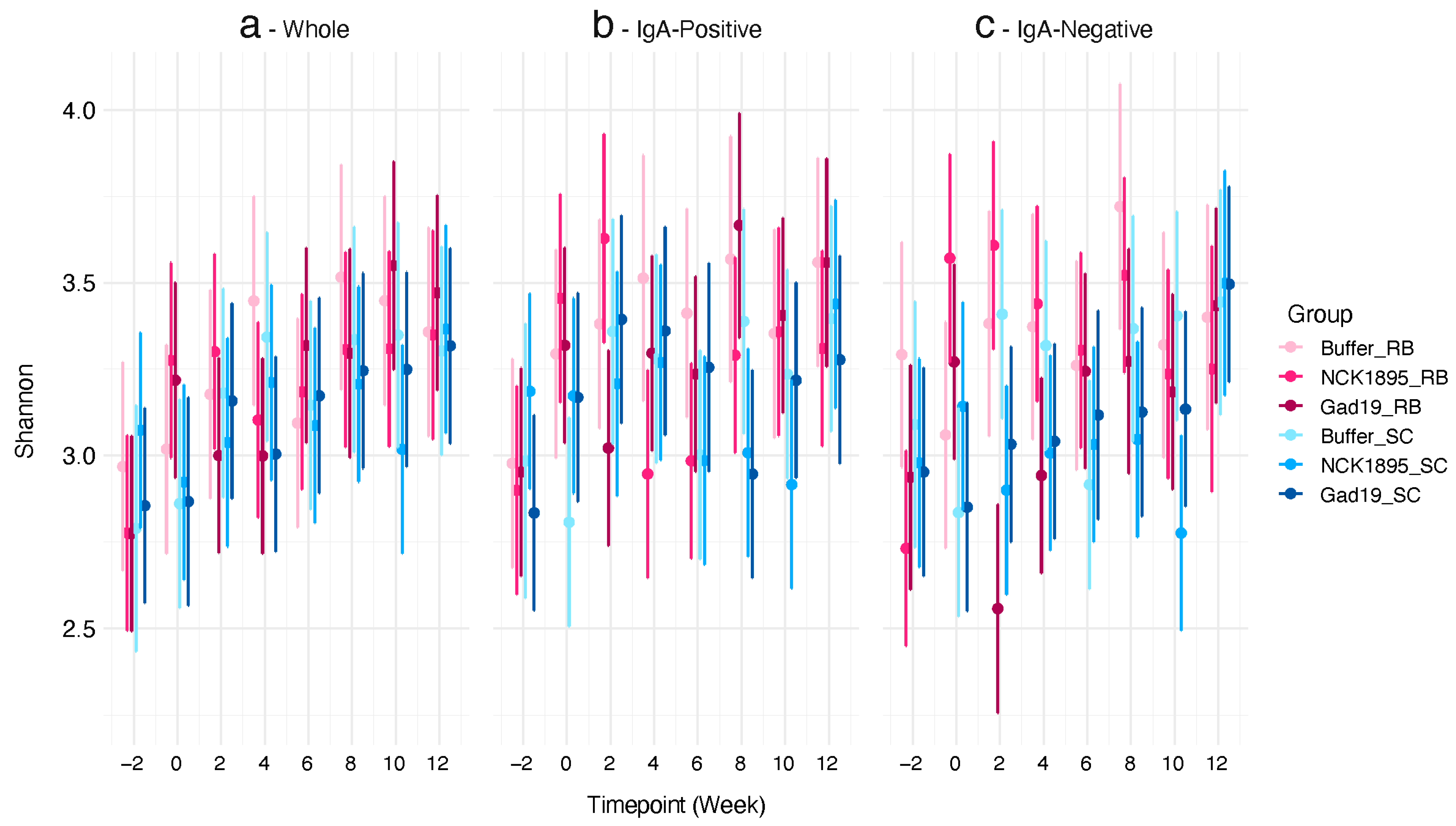
3.2. Impact on Alpha Diversity
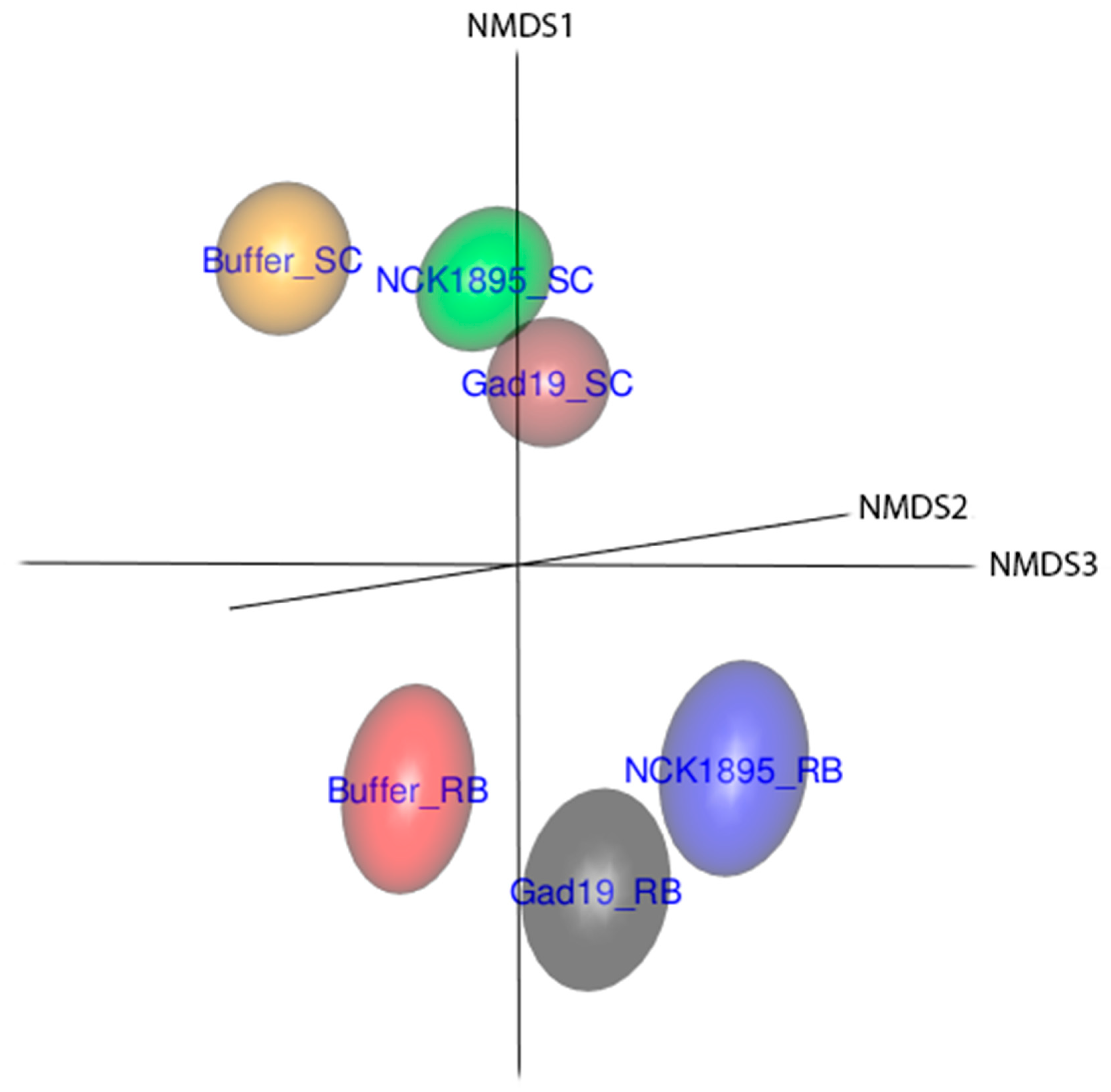
3.3. Temporal Changes in Beta Diversity
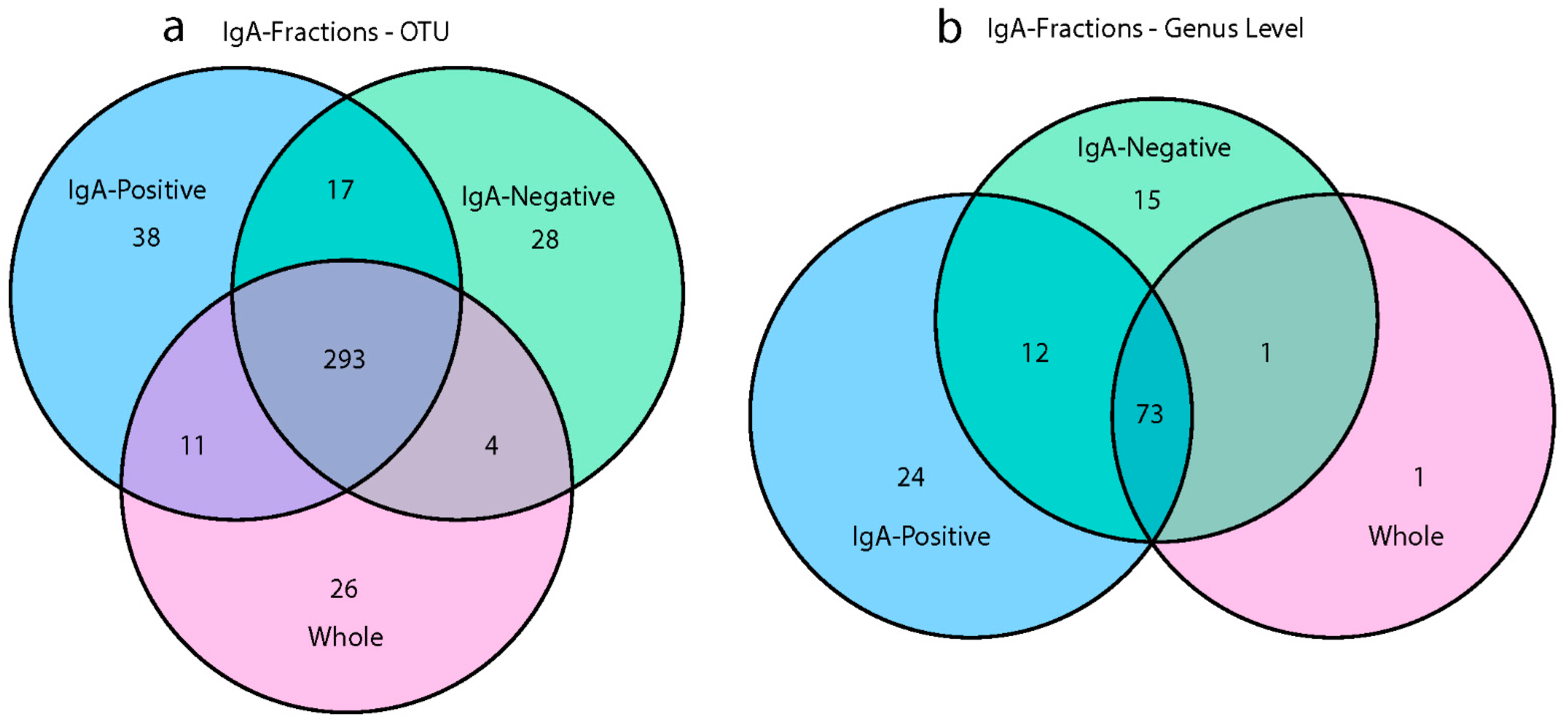
3.4. IgA Fractions Uncovered Low-Abundant Taxa
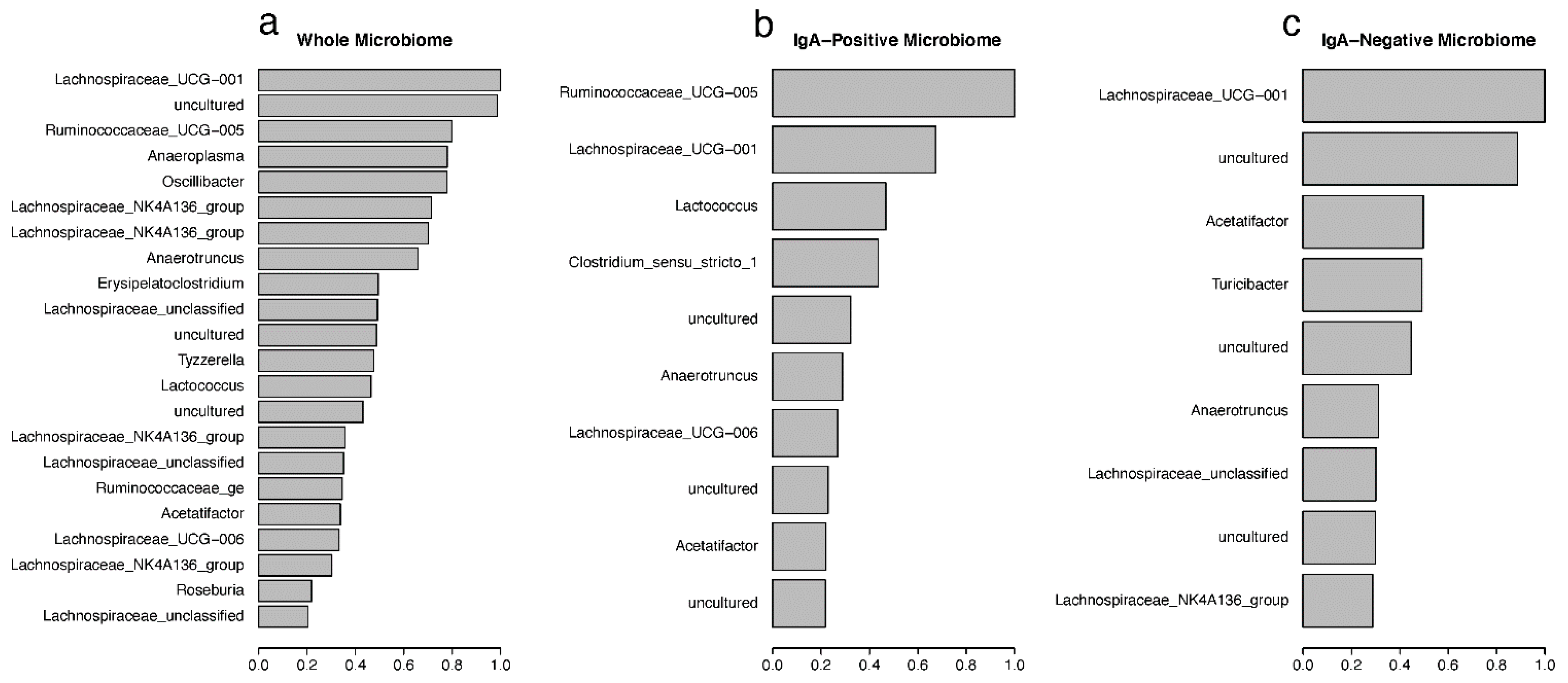
3.5. Random Forest Predictions of Important Taxa
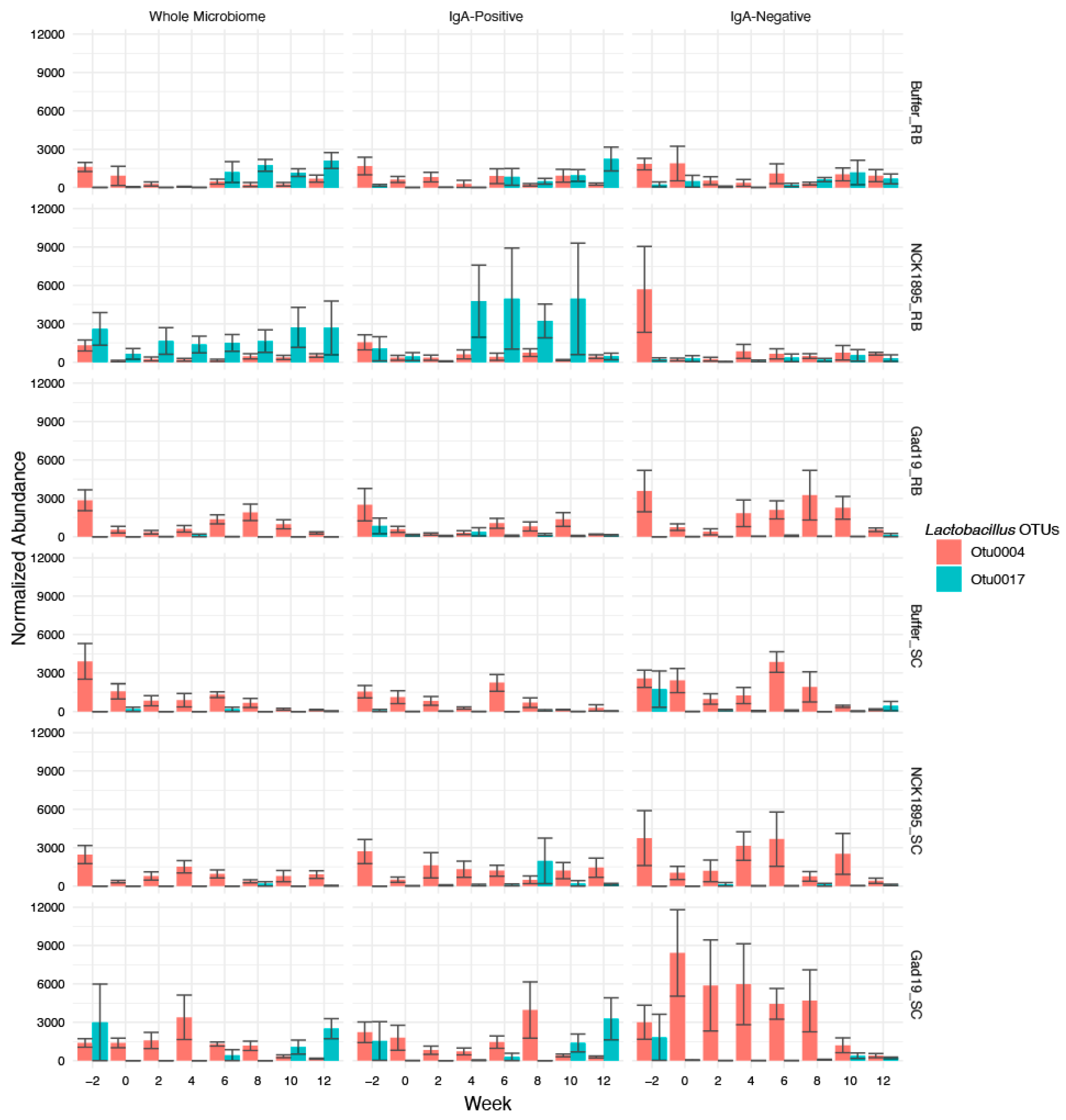
3.6. Lactobacilli Abundance and Importance over Time
3.7. Correlation between Antigen-Specific IgA and Microbial Taxa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kang, S.; Hong, S.; Lee, Y.-K.; Cho, S. Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting. Polymers 2018, 10, 948. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Kozlowski, P.A. Mucosal Vaccines: The Promise and the Challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, B.; Fischer, N.; Sansonetti, P.J. Mucosal Physical and Chemical Innate Barriers: Lessons from Microbial Evasion Strategies. Semin. Immunol. 2015, 27, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, F.; Wu, T.; Tang, H.; Zhao, Z. The Acid, Bile Tolerance and Antimicrobial Property of Lactobacillus Acidophilus NIT. Food Control. 2009, 20, 598–602. [Google Scholar] [CrossRef]
- Kajikawa, A.; Zhang, L.; LaVoy, A.; Bumgardner, S.; Klaenhammer, T.R.; Dean, G.A. Mucosal Immunogenicity of Genetically Modified Lactobacillus Acidophilus Expressing an HIV-1 Epitope within the Surface Layer Protein. PLoS ONE 2015, 10, e0141713. [Google Scholar] [CrossRef]
- Zeuthen, L.H.; Fink, L.N.; Frøkiaer, H. Toll-like Receptor 2 and Nucleotide-Binding Oligomerization Domain-2 Play Divergent Roles in the Recognition of Gut-Derived Lactobacilli and Bifidobacteria in Dendritic Cells. Immunology 2008, 124, 489–502. [Google Scholar] [CrossRef]
- Matsuguchi, T.; Takagi, A.; Matsuzaki, T.; Nagaoka, M.; Ishikawa, K.; Yokokura, T.; Yoshikai, Y. Lipoteichoic Acids from Lactobacillus Strains Elicit Strong Tumor Necrosis Factor Alpha-Inducing Activities in Macrophages through Toll-Like Receptor 2. CVI 2003, 10, 259–266. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 Is a General Sensor of Peptidoglycan through Muramyl Dipeptide (MDP) Detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef]
- Rhee, J.H. Current and New Approaches for Mucosal Vaccine Delivery. In Mucosal Vaccines; Elsevier: Amsterdam, The Netherlands, 2020; pp. 325–356. ISBN 978-0-12-811924-2. [Google Scholar]
- Shakya, A.K.; Chowdhury, M.Y.E.; Tao, W.; Gill, H.S. Mucosal Vaccine Delivery: Current State and a Pediatric Perspective. J. Control. Release 2016, 240, 394–413. [Google Scholar] [CrossRef]
- Kajikawa, A.; Zhang, L.; Long, J.; Nordone, S.; Stoeker, L.; LaVoy, A.; Bumgardner, S.; Klaenhammer, T.; Dean, G. Construction and Immunological Evaluation of Dual Cell Surface Display of HIV-1 Gag and Salmonella Enterica Serovar Typhimurium FliC in Lactobacillus Acidophilus for Vaccine Delivery. Clin. Vaccine Immunol. 2012, 19, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Bumgardner, S.A.; Zhang, L.; LaVoy, A.S.; Andre, B.; Frank, C.B.; Kajikawa, A.; Klaenhammer, T.R.; Dean, G.A. Nod2 Is Required for Antigen-Specific Humoral Responses against Antigens Orally Delivered Using a Recombinant Lactobacillus Vaccine Platform. PLoS ONE 2018, 13, 23. [Google Scholar] [CrossRef]
- Azegami, T.; Yuki, Y.; Kiyono, H. Challenges in Mucosal Vaccines for the Control of Infectious Diseases. Int. Immunol. 2014, 26, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Hoces, D.; Arnoldini, M.; Diard, M.; Loverdo, C.; Slack, E. Growing, Evolving and Sticking in a Flowing Environment: Understanding IgA Interactions with Bacteria in the Gut. Immunology 2020, 159, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Vogelzang, A.; Maruya, M.; Miyajima, M.; Murata, M.; Son, A.; Kuwahara, T.; Tsuruyama, T.; Yamada, S.; Matsuura, M.; et al. IgA Regulates the Composition and Metabolic Function of Gut Microbiota by Promoting Symbiosis between Bacteria. J. Exp. Med. 2018, 215, 2019–2034. [Google Scholar] [CrossRef]
- Okai, S.; Usui, F.; Yokota, S.; Hori-i, Y.; Hasegawa, M.; Nakamura, T.; Kurosawa, M.; Okada, S.; Yamamoto, K.; Nishiyama, E.; et al. High-Affinity Monoclonal IgA Regulates Gut Microbiota and Prevents Colitis in Mice. Nat. Microbiol. 2016, 1, 16103. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.J.; Bendelac, A. IgA Responses to Microbiota. Immunity 2018, 49, 211–224. [Google Scholar] [CrossRef]
- Bunker, J.J.; Erickson, S.A.; Flynn, T.M.; Henry, C.; Koval, J.C.; Meisel, M.; Jabri, B.; Antonopoulos, D.A.; Wilson, P.C.; Bendelac, A. Natural Polyreactive IgA Antibodies Coat the Intestinal Microbiota. Science 2017, 358, eaan6619. [Google Scholar] [CrossRef]
- Kau, A.L.; Planer, J.D.; Liu, J.; Rao, S.; Yatsunenko, T.; Trehan, I.; Manary, M.J.; Liu, T.-C.; Stappenbeck, T.S.; Maleta, K.M.; et al. Functional Characterization of IgA-Targeted Bacterial Taxa from Undernourished Malawian Children That Produce Diet-Dependent Enteropathy. Sci. Transl. Med. 2015, 7, 276ra24. [Google Scholar] [CrossRef]
- Abdo, Z.; LeCureux, J.; LaVoy, A.; Eklund, B.; Ryan, E.P.; Dean, G.A. Impact of Oral Probiotic Lactobacillus Acidophilus Vaccine Strains on the Immune Response and Gut Microbiome of Mice. PLoS ONE 2019, 14, 23. [Google Scholar] [CrossRef]
- Duong, T.; Miller, M.J.; Barrangou, R.; Azcarate-Peril, M.A.; Klaenhammer, T.R. Construction of Vectors for Inducible and Constitutive Gene Expression in Lactobacillus. Microb Biotechnol. 2011, 4, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Frey, A.; Di Canzio, J.; Zurakowski, D. A Statistically Defined Endpoint Titer Determination Method for Immunoassays. J. Immunol. Methods 1998, 221, 35–41. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 May 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. AEM 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Vegan: Community Ecology Package. R Package Version 2.4-3; 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 13 May 2019).
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential Abundance Analysis for Microbial Marker-Gene Surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Breiman, L.; Cutler, A. Breiman and Cutler’s Random Forests for Classification and Regression: Package ‘RandomForest’ version 4.6–14; 2018. Available online: http://cran.r-project.org/web/packages/randomForest/ (accessed on 13 May 2019).
- Hastie, T.; Friedman, J.H.; Tibshirani, R. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: New York, NY, USA, 2009; ISBN 9780387216065. [Google Scholar]
- D’Auria, G.; Peris-Bondia, F.; Džunková, M.; Mira, A.; Collado, M.C.; Latorre, A.; Moya, A. Active and Secreted IgA-Coated Bacterial Fractions from the Human Gut Reveal an under-Represented Microbiota Core. Sci. Rep. 2013, 3, 3515. [Google Scholar] [CrossRef]
- Jalanka-Tuovinen, J.; Salonen, A.; Nikkilä, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; de Vos, W.M. Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS ONE 2011, 6, e23035. [Google Scholar] [CrossRef]
- Kirkpatrick, B.D.; McKenzie, R.; O’Neill, J.P.; Larsson, C.J.; Bourgeois, A.L.; Shimko, J.; Bentley, M.; Makin, J.; Chatfield, S.; Hindle, Z.; et al. Evaluation of Salmonella Enterica Serovar Typhi (Ty2 AroC-SsaV-) M01ZH09, with a Defined Mutation in the Salmonella Pathogenicity Island 2, as a Live, Oral Typhoid Vaccine in Human Volunteers. Vaccine 2006, 24, 116–123. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, M.; Li, Z.; Frankel, F.R. Vaginal Protection and Immunity after Oral Immunization of Mice with a Novel Vaccine Strain of Listeria Monocytogenes Expressing Human Immunodeficiency Virus Type 1 Gag. JVI 2006, 80, 8880–8890. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.J.; Kumar, A.; Barnett, B.; Dow, S.W.; Ryan, E.P. Consumption of Rice Bran Increases Mucosal Immunoglobulin A Concentrations and Numbers of Intestinal Lactobacillus spp. J. Med. Food 2012, 15, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Scholtens, P.A.M.J.; Alliet, P.; Raes, M.; Alles, M.S.; Kroes, H.; Boehm, G.; Knippels, L.M.J.; Knol, J.; Vandenplas, Y. Fecal Secretory Immunoglobulin A Is Increased in Healthy Infants Who Receive a Formula with Short-Chain Galacto-Oligosaccharides and Long-Chain Fructo-Oligosaccharides. J. Nutr. 2008, 138, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Vandenplas, Y.; Greef, E.D.; Veereman, G. Prebiotics in Infant Formula. Gut Microbes 2014, 5, 681–687. [Google Scholar] [CrossRef]
- Desselberger, U. Differences of Rotavirus Vaccine Effectiveness by Country: Likely Causes and Contributing Factors. Pathogens 2017, 6, 65. [Google Scholar] [CrossRef]
- Parker, E.P.; Ramani, S.; Lopman, B.A.; Church, J.A.; Iturriza-Gómara, M.; Prendergast, A.J.; Grassly, N.C. Causes of Impaired Oral Vaccine Efficacy in Developing Countries. Future Microbiol. 2018, 13, 97–118. [Google Scholar] [CrossRef]
- Korpe, P.S.; Petri, W.A. Environmental Enteropathy: Critical Implications of a Poorly Understood Condition. Trends Mol. Med. 2012, 18, 328–336. [Google Scholar] [CrossRef]
- Uddin, M.I.; Islam, S.; Nishat, N.S.; Hossain, M.; Rafique, T.A.; Rashu, R.; Hoq, M.R.; Zhang, Y.; Saha, A.; Harris, J.B.; et al. Biomarkers of Environmental Enteropathy Are Positively Associated with Immune Responses to an Oral Cholera Vaccine in Bangladeshi Children. PLoS Negl. Trop. Dis. 2016, 10, e0005039. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally Rare Taxa Disproportionately Contribute to Temporal Changes in Microbial Diversity. mBio 2014, 5, e01371-14. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. BioMed. Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef] [Green Version]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where Less May Be More: How the Rare Biosphere Pulls Ecosystems Strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.E.; Palm, N.W. Functional Classification of the Gut Microbiota: The Key to Cracking the Microbiota Composition Code: Functional Classifications of the Gut Microbiota Reveal Previously Hidden Contributions of Indigenous Gut Bacteria to Human Health and Disease. BioEssays 2017, 39, 1700032. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Beasley, D.E.; Heděnec, P.; Xiao, Z.; Zhang, S.; Li, J.; Lin, Q.; Li, X. Diet Diversity Is Associated with Beta but Not Alpha Diversity of Pika Gut Microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, L.; Saltzman, E.; Thomsen, M.; Nikov, T.; Hall, S. Adjuvant Probiotics and the Intestinal Microbiome: Enhancing Vaccines and Immunotherapy Outcomes. Vaccines 2017, 5, 50. [Google Scholar] [CrossRef]
- Grazul, H.; Kanda, L.L.; Gondek, D. Impact of Probiotic Supplements on Microbiome Diversity Following Antibiotic Treatment of Mice. Gut Microbes 2016, 7, 101–114. [Google Scholar] [CrossRef]
- Derrien, M.; van Hylckama Vlieg, J.E.T. Fate, Activity, and Impact of Ingested Bacteria within the Human Gut Microbiota. Trends Microbiol. 2015, 23, 354–366. [Google Scholar] [CrossRef]
- Gerritsen, J.; Smidt, H.; Rijkers, G.T.; de Vos, W.M. Intestinal Microbiota in Human Health and Disease: The Impact of Probiotics. Genes Nutr. 2011, 6, 209–240. [Google Scholar] [CrossRef]
- Dassi, E.; Ferretti, P.; Covello, G.; Bertorelli, R.; Denti, M.A.; De Sanctis, V.; Tett, A.; Segata, N. The Short-Term Impact of Probiotic Consumption on the Oral Cavity Microbiome. Sci. Rep. 2018, 8, 10476. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Smith-Brown, P.; Morrison, M.; Krause, L.; Davies, P.S.W. Dairy and Plant Based Food Intakes Are Associated with Altered Faecal Microbiota in 2 to 3 Year Old Australian Children. Sci. Rep. 2016, 6, 32385. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-R.; Chou, T.-S.; Huang, C.-Y.; Hsiao, J.-K. A Potential Probiotic- Lachnospiraceae NK4A136 Group: Evidence from the Restoration of the Dietary Pattern from a High-Fat Diet. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Menni, C. Gut Microbiome Diversity and High-Fibre Intake Are Related to Lower Long-Term Weight Gain. Int. J. Obes. 2017, 41, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, Z.; Yu, K.; Ding, R.; Ye, K.; Dai, C.; Xu, X.; Zhou, G.; Li, C. High-Salt Diet Has a Certain Impact on Protein Digestion and Gut Microbiota: A Sequencing and Proteome Combined Study. Front. Microbiol. 2017, 8, 1838. [Google Scholar] [CrossRef] [PubMed]
- Byerley, L.O.; Samuelson, D.; Blanchard, E.; Luo, M.; Lorenzen, B.N.; Banks, S.; Ponder, M.A.; Welsh, D.A.; Taylor, C.M. Changes in the Gut Microbial Communities Following Addition of Walnuts to the Diet. J. Nutr. Biochem. 2017, 48, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Morrison, M.; Cao, K.-A.L.; Pruilh, S.; Davies, P.S.W.; Wall, C.; Lovell, A.; Hill, R.J. Dietary Intake Influences Gut Microbiota Development of Healthy Australian Children from the Age of One to Two Years. Sci. Rep. 2019, 9, 12476. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Treatment | Diet |
|---|---|---|---|
| Buffer_SC | 7 | Carrier buffer only | Standard chow |
| NCK1895_SC | 8 | NCK1895 | Standard chow |
| GAD19_SC | 8 | GAD19 | Standard chow |
| Buffer_RB | 8 | Carrier buffer only | 10% Rice bran |
| NCK1895_RB | 8 | NCK1895 | 10% Rice bran |
| GAD19_RB | 8 | GAD19 | 10% Rice bran |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fox, B.E.; Vilander, A.C.; Gilfillan, D.; Dean, G.A.; Abdo, Z. Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome. Vaccines 2022, 10, 1465. https://doi.org/10.3390/vaccines10091465
Fox BE, Vilander AC, Gilfillan D, Dean GA, Abdo Z. Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome. Vaccines. 2022; 10(9):1465. https://doi.org/10.3390/vaccines10091465
Chicago/Turabian StyleFox, Bridget E., Allison C. Vilander, Darby Gilfillan, Gregg A. Dean, and Zaid Abdo. 2022. "Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome" Vaccines 10, no. 9: 1465. https://doi.org/10.3390/vaccines10091465
APA StyleFox, B. E., Vilander, A. C., Gilfillan, D., Dean, G. A., & Abdo, Z. (2022). Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome. Vaccines, 10(9), 1465. https://doi.org/10.3390/vaccines10091465