
Characterization of Live-Attenuated Powassan Virus Vaccine Candidates Identifies an Efficacious Prime-Boost Strategy for Mitigating Powassan Virus Disease in a Murine Model
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Construction
2.2.1. Generation of a Full-Length DTV Infectious Clone
2.2.2. Construction of Infectious Clones for Recoded DTV with Increased CpG and UpA Frequencies
2.2.3. Generation of Three Chimeric YFV-17D-DTV Infectious Clones
Generation of Chimera I Containing a Mutation of the YFV NS1 N-Linked Glycosylation Site (CIGM)
2.2.4. Generation of Plasmid Used for POWV RNA Standard
2.3. Virus Production and Titration
2.4. POWV EDIII Expression and Purification
2.5. Vaccination Schemes
2.6. Mouse Infections and Monitoring/Scoring
2.7. Tissue Harvest, Virus Inactivation, and RNA Extraction
2.8. Generation of RNA for Use as RT-qPCR Standards
2.9. Quantification of Genome Copies by RT-qPCR for YFV and DTV
2.10. Sequence Analysis of Viral RNA Isolated from Brain
3. Results
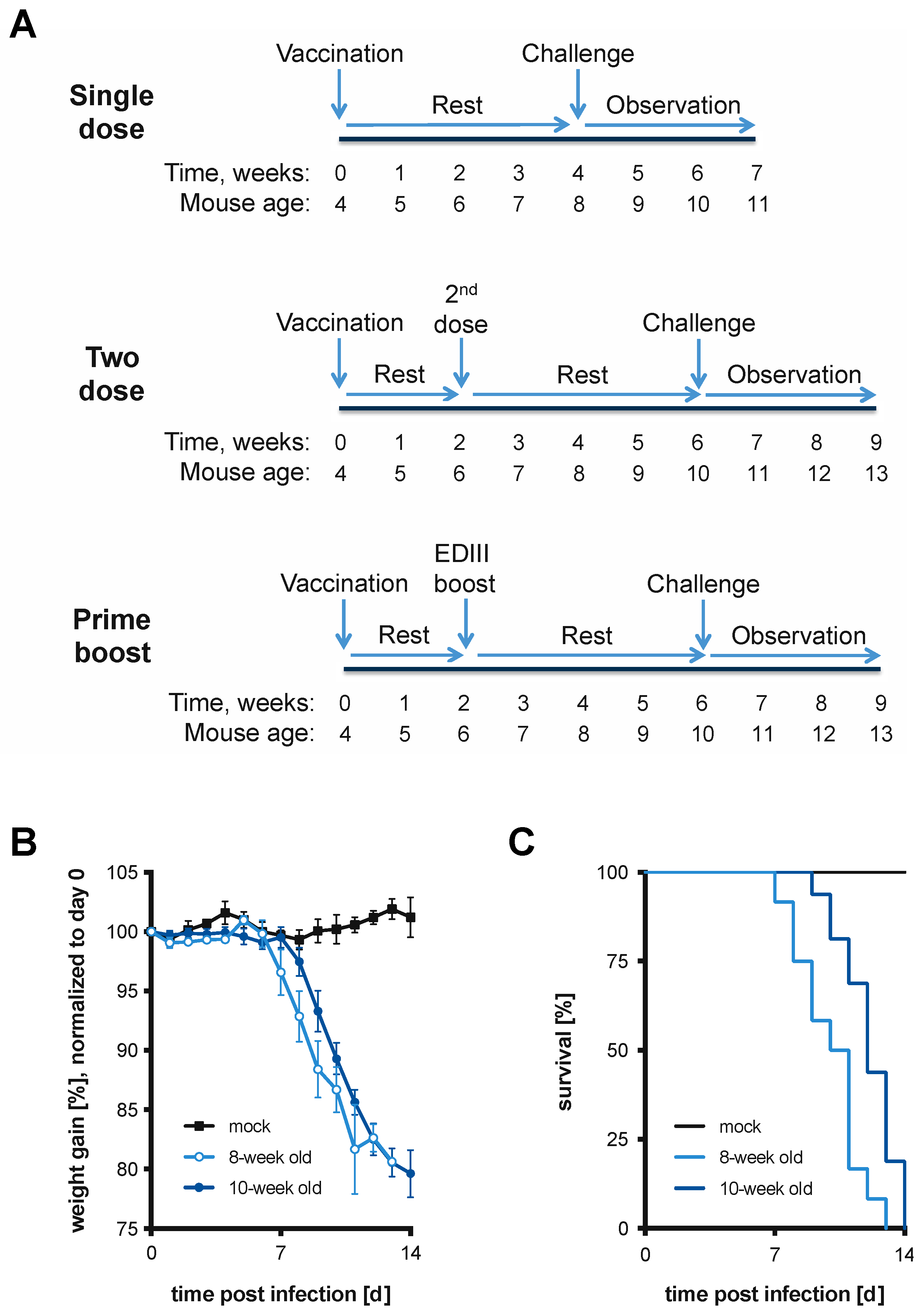
3.1. Lethal Model of DTV Infection in 8- and 10-Week-Old BL6 Mice
3.2. CpG and UpA Recoded DTV Viruses
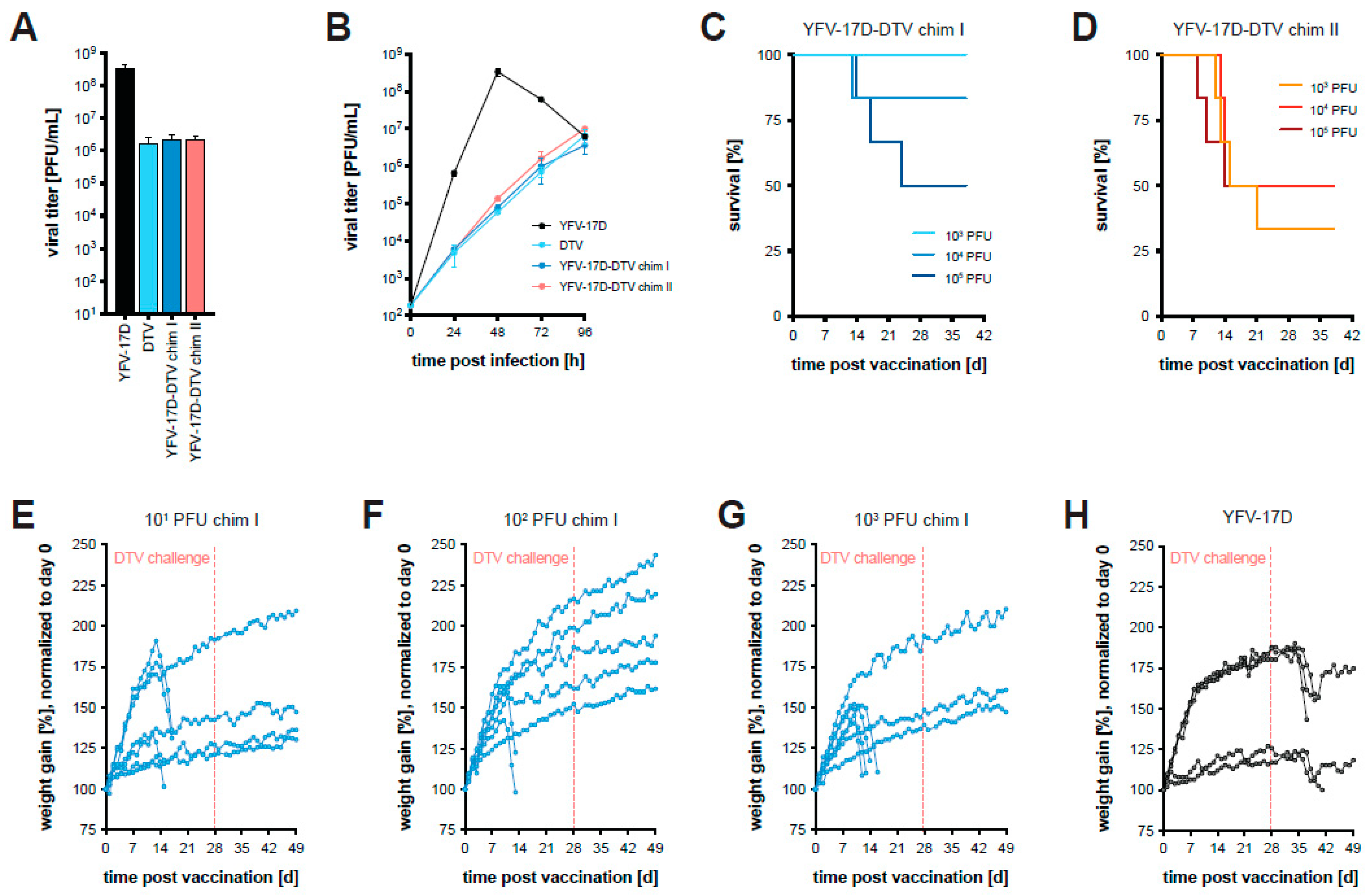
3.3. Chimeric YFV-17D-DTV Protects against DTV In Vivo
3.4. Removal of a Glycosylation Motif within YFV NS1 Further Attenuates Chimera I In Vitro
3.5. Chimera I Glycosylation Mutant Partially Protects In Vivo against Lethal DTV Challenge
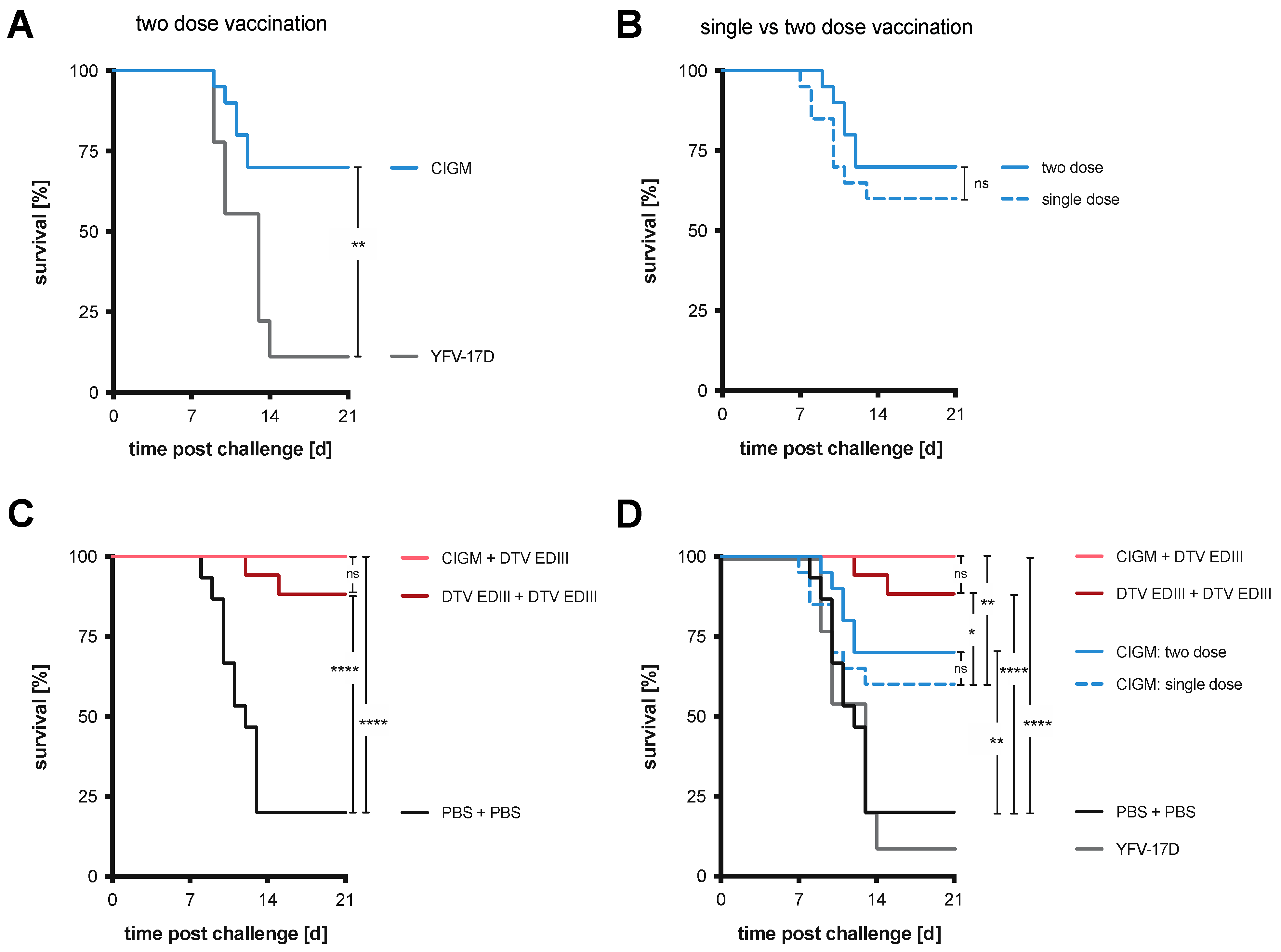
3.6. Chimera I Glycosylation Mutant Tested in Two-Dose and Prime-Boost Regimens
3.7. Differences in DTV Levels Post-Challenge after Vaccination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hermance, M.E.; Thangamani, S. Powassan Virus: An Emerging Arbovirus of Public Health Concern in North America. Vector Borne Zoonotic Dis. 2017, 17, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, R.; Lindsey, N.P.; Fischer, M.; Gregory, C.J.; Hinckley, A.F.; Mead, P.S.; Paz-Bailey, G.; Waterman, S.H.; Drexler, N.A.; Kersh, G.J.; et al. Vital Signs: Trends in Reported Vectorborne Disease Cases—United States and Territories, 2004-2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, C.B.; Eisen, L.; Eisen, R.J. The Rise of Ticks and Tickborne Diseases in the United States-Introduction. J. Med. Entomol. 2021, 58, 1487–1489. [Google Scholar] [CrossRef]
- Anderson, J.F.; Armstrong, P.M. Prevalence and genetic characterization of Powassan virus strains infecting Ixodes scapularis in Connecticut. Am. J. Trop. Med. Hyg. 2012, 87, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goethert, H.K.; Mather, T.N.; Johnson, R.W.; Telford, S.R., 3rd. Incrimination of shrews as a reservoir for Powassan virus. Commun. Biol. 2021, 4, 1319. [Google Scholar] [CrossRef]
- Brackney, D.E.; Brown, I.K.; Nofchissey, R.A.; Fitzpatrick, K.A.; Ebel, G.D. Homogeneity of Powassan virus populations in naturally infected Ixodes scapularis. Virology 2010, 402, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Pesko, K.N.; Torres-Perez, F.; Hjelle, B.L.; Ebel, G.D. Molecular epidemiology of Powassan virus in North America. J. Gen. Virol. 2010, 91, 2698–2705. [Google Scholar] [CrossRef]
- VanBlargan, L.A.; Himansu, S.; Foreman, B.M.; Ebel, G.D.; Pierson, T.C.; Diamond, M.S. An mRNA Vaccine Protects Mice against Multiple Tick-Transmitted Flavivirus Infections. Cell Rep. 2018, 25, 3382–3392.e3383. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Kudchodkar, S.B.; Ho, M.; Reuschel, E.L.; Reynolds, E.; Xu, Z.; Bordoloi, D.; Ugen, K.E.; Tebas, P.; Kim, J.; et al. A novel synthetic DNA vaccine elicits protective immune responses against Powassan virus. PLoS Negl. Trop. Dis. 2020, 14, e0008788. [Google Scholar] [CrossRef]
- Stone, E.T.; Hassert, M.; Geerling, E.; Wagner, C.; Brien, J.D.; Ebel, G.D.; Hirsch, A.J.; German, C.; Smith, J.L.; Pinto, A.K. Balanced T and B cell responses are required for immune protection against Powassan virus in virus-like particle vaccination. Cell Rep. 2022, 38, 110388. [Google Scholar] [CrossRef]
- Malonis, R.J.; Georgiev, G.I.; Haslwanter, D.; VanBlargan, L.A.; Fallon, G.; Vergnolle, O.; Cahill, S.M.; Harris, R.; Cowburn, D.; Chandran, K.; et al. A Powassan virus domain III nanoparticle immunogen elicits neutralizing and protective antibodies in mice. PLoS Pathog. 2022, 18, e1010573. [Google Scholar] [CrossRef] [PubMed]
- Meagher, J.L.; Takata, M.; Goncalves-Carneiro, D.; Keane, S.C.; Rebendenne, A.; Ong, H.; Orr, V.K.; MacDonald, M.R.; Stuckey, J.A.; Bieniasz, P.D.; et al. Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. USA 2019, 116, 24303–24309. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ma, J.; Sun, J.; Gao, G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, X.; Goff, S.P.; Gao, G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012, 31, 4236–4246. [Google Scholar] [CrossRef]
- Goncalves-Carneiro, D.; Mastrocola, E.; Lei, X.; DaSilva, J.; Chan, Y.F.; Bieniasz, P.D. Rational attenuation of RNA viruses with zinc finger antiviral protein. Nat. Microbiol. 2022, 7, 1558–1567. [Google Scholar] [CrossRef]
- Fros, J.J.; Dietrich, I.; Alshaikhahmed, K.; Passchier, T.C.; Evans, D.J.; Simmonds, P. CpG and UpA dinucleotides in both coding and non-coding regions of echovirus 7 inhibit replication initiation post-entry. eLife 2017, 6, e29112. [Google Scholar] [CrossRef] [Green Version]
- Pulendran, B.; Ahmed, R. Immunological mechanisms of vaccination. Nat. Immunol. 2011, 12, 509–517. [Google Scholar] [CrossRef]
- Wieten, R.W.; Jonker, E.F.; van Leeuwen, E.M.; Remmerswaal, E.B.; Ten Berge, I.J.; de Visser, A.W.; van Genderen, P.J.; Goorhuis, A.; Visser, L.G.; Grobusch, M.P.; et al. A Single 17D Yellow Fever Vaccination Provides Lifelong Immunity; Characterization of Yellow-Fever-Specific Neutralizing Antibody and T-Cell Responses after Vaccination. PLoS ONE 2016, 11, e0149871. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.S.; Dalrymple, J.M.; Strauss, J.H.; Rice, C.M. Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc. Natl. Acad. Sci. USA 1987, 84, 2019–2023. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.D.T. Yellow fever live attenuated vaccine: A very successful live attenuated vaccine but still we have problems controlling the disease. Vaccine 2017, 35, 5951–5955. [Google Scholar] [CrossRef]
- Poland, J.D.; Calisher, C.H.; Monath, T.P.; Downs, W.G.; Murphy, K. Persistence of neutralizing antibody 30–35 years after immunization with 17D yellow fever vaccine. Bull. World Health Organ. 1981, 59, 895–900. [Google Scholar]
- Akondy, R.S.; Monson, N.D.; Miller, J.D.; Edupuganti, S.; Teuwen, D.; Wu, H.; Quyyumi, F.; Garg, S.; Altman, J.D.; Del Rio, C.; et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 2009, 183, 7919–7930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, E.A.; LaFond, R.E.; Gates, T.J.; Mai, D.T.; Malhotra, U.; Kwok, W.W. Yellow fever vaccination elicits broad functional CD4+ T cell responses that recognize structural and nonstructural proteins. J. Virol. 2013, 87, 12794–12804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, M.R.; Kongsgaard, M.; Steffensen, M.A.; Fenger, C.; Rasmussen, M.; Skjodt, K.; Finsen, B.; Stryhn, A.; Buus, S.; Christensen, J.P.; et al. CD8+ T cells complement antibodies in protecting against yellow fever virus. J. Immunol. 2015, 194, 1141–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jia, R.; Shen, H.; Wang, M.; Yin, Z.; Cheng, A. Structures and Functions of the Envelope Glycoprotein in Flavivirus Infections. Viruses 2017, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–13100. [Google Scholar] [CrossRef] [Green Version]
- Crill, W.D.; Roehrig, J.T. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J. Virol. 2001, 75, 7769–7773. [Google Scholar] [CrossRef] [Green Version]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef]
- Appaiahgari, M.B.; Vrati, S. IMOJEV((R)): A Yellow fever virus-based novel Japanese encephalitis vaccine. Expert. Rev. Vaccines 2010, 9, 1371–1384. [Google Scholar] [CrossRef]
- Tully, D.; Griffiths, C.L. Dengvaxia: The world’s first vaccine for prevention of secondary dengue. Ther. Adv. Vaccines Immunother. 2021, 9, 25151355211015839. [Google Scholar] [CrossRef]
- Seino, K.K.; Long, M.T.; Gibbs, E.P.; Bowen, R.A.; Beachboard, S.E.; Humphrey, P.P.; Dixon, M.A.; Bourgeois, M.A. Comparative efficacies of three commercially available vaccines against West Nile Virus (WNV) in a short-duration challenge trial involving an equine WNV encephalitis model. Clin. Vaccine Immunol. 2007, 14, 1465–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, J.L.; Anishchenko, M.; Hermance, M.; Romo, H.; Chen, C.I.; Thangamani, S.; Brault, A.C. Generation of a Lineage II Powassan Virus (Deer Tick Virus) cDNA Clone: Assessment of Flaviviral Genetic Determinants of Tick and Mosquito Vector Competence. Vector Borne Zoonotic Dis. 2018, 18, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Fros, J.J.; Goonawardane, N.; Dietrich, I.; Ibrahim, A.; Alshaikhahmed, K.; Nguyen, D.; Simmonds, P. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res. 2019, 47, 8061–8083. [Google Scholar] [CrossRef]
- Esswein, S.R.; Gristick, H.B.; Jurado, A.; Peace, A.; Keeffe, J.R.; Lee, Y.E.; Voll, A.V.; Saeed, M.; Nussenzweig, M.C.; Rice, C.M.; et al. Structural basis for Zika envelope domain III recognition by a germline version of a recurrent neutralizing antibody. Proc. Natl. Acad. Sci. USA 2020, 117, 9865–9875. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bozzacco, L.; Keeffe, J.R.; Khouri, R.; Olsen, P.C.; Gazumyan, A.; Schaefer-Babajew, D.; Avila-Rios, S.; Nogueira, L.; Patel, R.; et al. Recurrent Potent Human Neutralizing Antibodies to Zika Virus in Brazil and Mexico. Cell 2017, 169, 597–609.e511. [Google Scholar] [CrossRef] [Green Version]
- Sapparapu, G.; Fernandez, E.; Kose, N.; Bin, C.; Fox, J.M.; Bombardi, R.G.; Zhao, H.; Nelson, C.A.; Bryan, A.L.; Barnes, T.; et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016, 540, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; de Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93, e00715-19. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.D.; Barrett, A.D. Live Attenuated Yellow Fever 17D Vaccine: A Legacy Vaccine Still Controlling Outbreaks In Modern Day. Curr. Infect. Dis. Rep. 2017, 19, 14. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. Degrees of maturity: The complex structure and biology of flaviviruses. Curr. Opin. Virol. 2012, 2, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moudy, R.M.; Zhang, B.; Shi, P.Y.; Kramer, L.D. West Nile virus envelope protein glycosylation is required for efficient viral transmission by Culex vectors. Virology 2009, 387, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Muthukrishnan, E.; Natarajan, S.K.; Steffen, D.; Vu, H.L.X.; Delhon, G.; Osorio, F.A.; Petro, T.M.; et al. Zika Virus Encoding Nonglycosylated Envelope Protein Is Attenuated and Defective in Neuroinvasion. J. Virol. 2017, 91, e01348-17. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Li, S.; Dong, F.; Zhang, Y.; Lin, Y.; Wang, J.; Zou, Z.; Zheng, A. N-glycosylation of Viral E Protein Is the Determinant for Vector Midgut Invasion by Flaviviruses. mBio 2018, 9, e00046-18. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef] [Green Version]
- Muylaert, I.R.; Chambers, T.J.; Galler, R.; Rice, C.M. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: Effects on virus replication and mouse neurovirulence. Virology 1996, 222, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Fros, J.J.; Visser, I.; Tang, B.; Yan, K.; Nakayama, E.; Visser, T.M.; Koenraadt, C.J.M.; van Oers, M.M.; Pijlman, G.P.; Suhrbier, A.; et al. The dinucleotide composition of the Zika virus genome is shaped by conflicting evolutionary pressures in mammalian hosts and mosquito vectors. PLoS Biol. 2021, 19, e3001201. [Google Scholar] [CrossRef]
- Goonawardane, N.; Nguyen, D.; Simmonds, P. Association of Zinc Finger Antiviral Protein Binding to Viral Genomic RNA with Attenuation of Replication of Echovirus 7. mSphere 2021, 6, e01138-20. [Google Scholar] [CrossRef]
- Gaunt, E.; Wise, H.M.; Zhang, H.; Lee, L.N.; Atkinson, N.J.; Nicol, M.Q.; Highton, A.J.; Klenerman, P.; Beard, P.M.; Dutia, B.M.; et al. Elevation of CpG frequencies in influenza A genome attenuates pathogenicity but enhances host response to infection. eLife 2016, 5, e12735. [Google Scholar] [CrossRef]
- Ficarelli, M.; Wilson, H.; Pedro Galao, R.; Mazzon, M.; Antzin-Anduetza, I.; Marsh, M.; Neil, S.J.; Swanson, C.M. KHNYN is essential for the zinc finger antiviral protein (ZAP) to restrict HIV-1 containing clustered CpG dinucleotides. eLife 2019, 8, e46767. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Sequence Composition | G + C Content | Total CpG (Change) | Total UpA (Change) | Ratio CpG | Ratio UpA |
|---|---|---|---|---|---|---|
| Full length | Native | 0.5274 | 380 | 281 | 0.51 | 0.46 |
| R1 * | WT | 0.5333 | 56 (-) | 49 (-) | 0.5231 | 0.5848 |
| CDLR | 0.5333 | 56 (0) | 49 (0) | 0.5231 | 0.5848 | |
| CpG-1 | 0.533 | 106 (+50) | 49 (0) | 0.9901 | 0.5848 | |
| CpG-2 | 0.5333 | 133 (+77) | 49 (0) | 1.2423 | 0.5848 | |
| UpA-1 | 0.5333 | 56 (0) | 83 (+34) | 0.5231 | 0.9907 | |
| UpA-2 | 0.5333 | 56 (0) | 115 (+66) | 0.5231 | 1.3726 | |
| R2 * | WT | 0.5213 | 54 | 33 | 0.5918 | 0.4238 |
| CDLR | 0.5213 | 54 | 33 | 0.5918 | 0.4238 | |
| CpG-1 | 0.5213 | 90 (+36) | 33 (0) | 0.9863 | 0.4238 | |
| CpG-2 | 0.5213 | 121 (+67) | 33 (0) | 1.326 | 0.4238 | |
| UpA-1 | 0.5213 | 54 (0) | 77 (+44) | 0.5918 | 0.989 | |
| UpA-2 | 0.5213 | 54 (0) | 100 (+67) | 0.5918 | 1.2844 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, A.M.; Yip, E.Z.; Ashbrook, A.W.; Goonawardane, N.; Quirk, C.; Rice, C.M.; MacDonald, M.R.; Hoffmann, H.-H. Characterization of Live-Attenuated Powassan Virus Vaccine Candidates Identifies an Efficacious Prime-Boost Strategy for Mitigating Powassan Virus Disease in a Murine Model. Vaccines 2023, 11, 612. https://doi.org/10.3390/vaccines11030612
Cheung AM, Yip EZ, Ashbrook AW, Goonawardane N, Quirk C, Rice CM, MacDonald MR, Hoffmann H-H. Characterization of Live-Attenuated Powassan Virus Vaccine Candidates Identifies an Efficacious Prime-Boost Strategy for Mitigating Powassan Virus Disease in a Murine Model. Vaccines. 2023; 11(3):612. https://doi.org/10.3390/vaccines11030612
Chicago/Turabian StyleCheung, Andrew M., Elaine Z. Yip, Alison W. Ashbrook, Niluka Goonawardane, Corrine Quirk, Charles M. Rice, Margaret R. MacDonald, and Hans-Heinrich Hoffmann. 2023. "Characterization of Live-Attenuated Powassan Virus Vaccine Candidates Identifies an Efficacious Prime-Boost Strategy for Mitigating Powassan Virus Disease in a Murine Model" Vaccines 11, no. 3: 612. https://doi.org/10.3390/vaccines11030612
APA StyleCheung, A. M., Yip, E. Z., Ashbrook, A. W., Goonawardane, N., Quirk, C., Rice, C. M., MacDonald, M. R., & Hoffmann, H.-H. (2023). Characterization of Live-Attenuated Powassan Virus Vaccine Candidates Identifies an Efficacious Prime-Boost Strategy for Mitigating Powassan Virus Disease in a Murine Model. Vaccines, 11(3), 612. https://doi.org/10.3390/vaccines11030612