Abstract
Tumor vaccine is a promising immunotherapy for solid tumors. Therapeutic tumor vaccines aim at inducing tumor regression, establishing durable antitumor memory, and avoiding non-specific or adverse reactions. However, tumor-induced immune suppression and immune resistance pose challenges to achieving this goal. In this article, we review multiple challenges currently faced in the development of therapeutic tumor vaccines, with a particular focus on anonymous antigen vaccines in situ as a new direction. We summarize the research progress in this area, aiming to provide a reference for future studies on tumor vaccines.
1. Introduction
Tumor immunotherapy refers to re-activating the immune system of patients to fight against tumors, in which immune cells play critical roles [1]. For an effective antitumor immune response to eliminate cancer cells, a series of sequential steps termed the “cancer- immunity cycle” must be initiated [2]. Within this cycle, antigen-presenting cells (APCs) first process antigens derived from tumor cells. Subsequently, dendritic cells (DCs) present these antigens on major histocompatibility complex (MHC) class I and II molecules to T cells, thereby initiating and activating effector T cells against tumor-specific antigens. Ultimately, activated effector T cells recognize and attack tumor cells through their T-cell receptors (TCRs) [2,3,4]. Several tumor immunotherapies have entered clinical application, such as immune checkpoint blockade (ICB) and chimeric antigen receptor T-cell (CAR-T) therapy, each targeting different stages of the cancer-immunity cycle [5,6]. However, the efficacy of first-generation immune checkpoint inhibitors (ICIs), like PD-1/PD-L1 antibodies, is limited by the infiltration of cytotoxic T lymphocytes (CTLs) into tumors [7,8]. Additionally, the clinical application of CAR-T therapy is restricted by high costs, time requirements, and adverse reactions [9]. The presence of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) at tumor sites would also lead to poor treatment outcomes. While these methods target the latter stages of the cancer-mmunity cycle, tumor vaccines focus on the upstream of this cycle by inducing CTLs and humoral immune responses through tumor antigens, and are therefore regarded as promising candidates for activating the immune system [10,11].
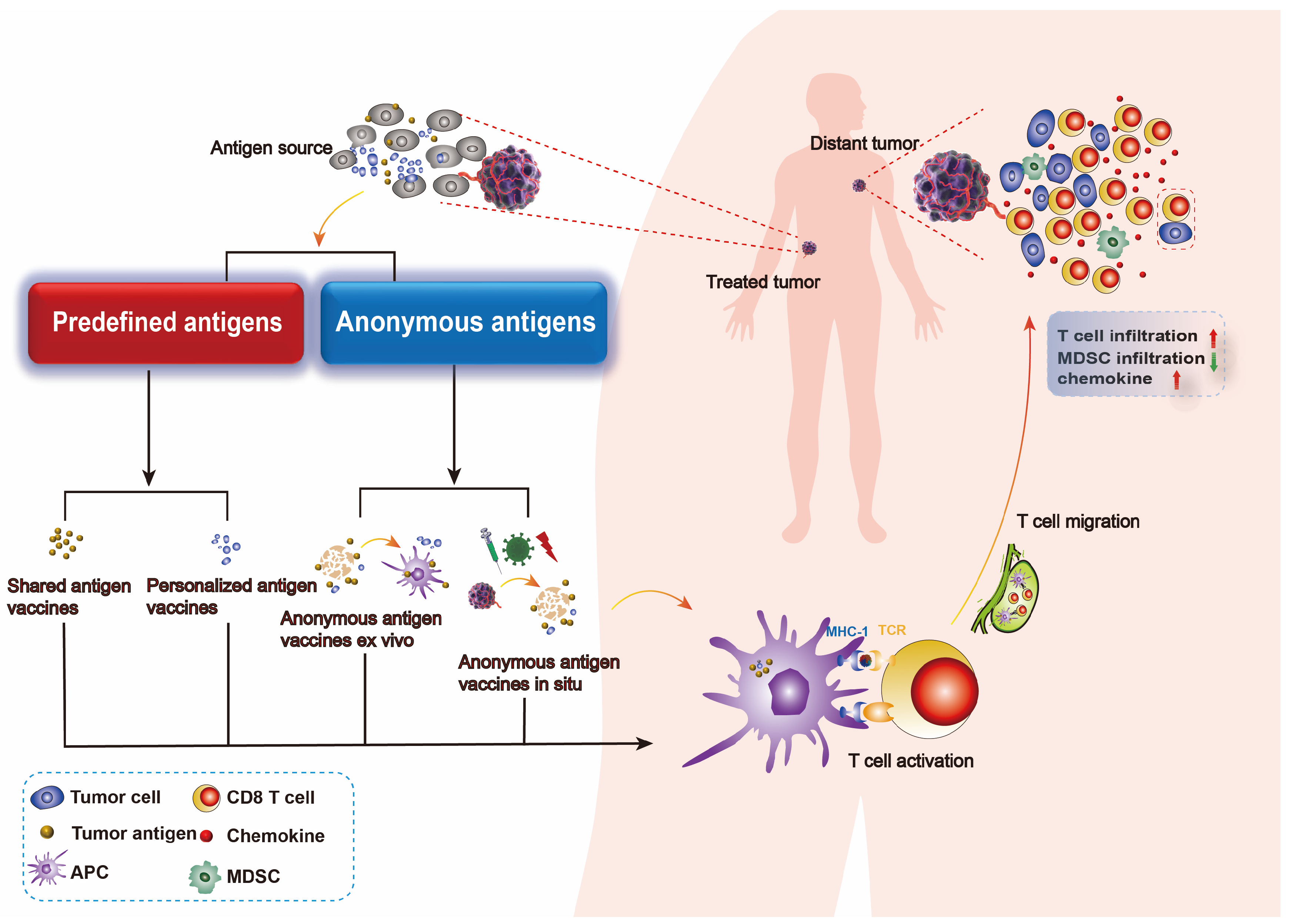
Tumor vaccine platforms can be classified into four types based on their preparation methods and sources of tumor antigen: cellular vaccines, viral vaccines, peptide vaccines, and nucleic acid vaccines [12,13]. Cell-based vaccines are the main form of original tumor vaccines, with DC vaccines showing significant results in clinical trials [14]. Viral vaccines utilize viruses as vectors to treat and prevent tumors. Peptide vaccines consist of known or predicted tumor antigen epitopes and typically exhibit lower immunogenicity, thus are combined with adjuvants to enhance their immunogenicity [15,16]. Nucleic acid vaccines include DNA and RNA vaccines composed of genes encoding pathogen antigens and vector groups [17]. DNA vaccines are closed circular DNA plasmids encoding tumor-associated antigens (TAAs) or immunomodulatory molecules to induce tumor-specific responses; mRNA vaccines are synthesized in vitro and can encode antigens post-internalization to stimulate immune responses [18]. Based on our understanding of tumor-specific immunogenic antigens, the number of patients expressing corresponding tumor-specific antigens, and how these antigens co-localize with professional APCs, tumor vaccines can further be divided into predefined (known) antigen vaccines and anonymous (unknown) antigen vaccines. Predefined antigen vaccines are categorized into shared antigen vaccines—expressed in multiple patients’ tumors—and personalized antigen vaccines tailored for individual patients. Shared antigen vaccines are prepared with antigens widely expressing in a group of patients, thus may not be effective for some patients lacking the targeted antigen. Differently, personalized antigen vaccines are prepared with antigens derived from a certain patient and therefore could be more specific and effective, which also requires higher costs. Anonymous antigen vaccines can be classified as anonymous antigen vaccines ex vivo or anonymous antigen vaccines in situ that co-localize with APCs either in vitro or at the tumor site [19] (Figure 1). Anonymous antigen vaccines ex vivo refer to the transfusion of autologous APCs which are stimulated by lysates of excised tumor tissues or cells ex vivo, while anonymous antigen vaccines in situ refer to vaccines that promote the release of tumor antigens in situ and activate endogenous APCs.

Figure 1.
Types of tumor vaccine. Tumor vaccines are prepared from tumor antigen, thereby they could activate specific antitumor immune responses, including an increase in chemokine expression and T-cell infiltration within the tumor and a decrease in MDSC infiltration. Different sources of tumor antigens draw a distinction between predefined antigen vaccines and anonymous antigen vaccines. Predefined antigen vaccines consist of shared antigen vaccines and personalized antigen vaccines, both of which require the preparation of a certain tumor antigen for the vaccine. Anonymous antigen vaccines consist of anonymous antigen vaccines ex vivo and anonymous antigen vaccines in vivo, which utilize all the tumor antigen profiles in a patient to act as a vaccine.
2. Predefined Antigen Vaccines and Challenges of Their Therapeutic Applications
2.1. Predefined Antigen Vaccines
In cancer immunotherapy, predefined antigen vaccines have shown promising results. They can be classified into shared and personalized antigens based on their expression across patient populations [19]. Shared antigens are those expressed in a certain proportion of patients, for which corresponding standard tests are utilized [20]. For example, the novel epitope TSA epidermal growth factor receptor variant III (EGFRvIII) is expressed in approximately 25% of glioblastomas [21]. Another notable example are the human papillomavirus (HPV) E6 and E7 proteins, which are found in about 60% of oropharyngeal cancers and nearly all cervical cancers [22]. These shared antigens can serve as targets for therapeutic vaccines that are applicable to multiple patients with similar tumor characteristics. In contrast, personalized neoantigens are unique to each patient and require custom preparation. A notable example is autogene cevumeran, an mRNA-based personalized cancer vaccine developed by BioNTech (Mainz, Germany) that targets up to 20 unique neoantigens identified from each patient’s tumor. Clinical trials have shown that this vaccine can induce durable polyfunctional neoantigen-specific effector CD8+ T-cell responses against pancreatic cancer. This personalized approach could elicit stronger antitumor immune responses while minimizing the risk of off-target effects [23]. Recent initiatives like the NHS’s personalized cancer vaccine program aim to tailor treatments based on individual patient profiles, enhancing specificity and effectiveness. The landscape of therapeutic cancer vaccines is evolving, with ongoing clinical trials examining their efficacy across various malignancies such as lung cancer, breast cancer, and melanoma [24]. Despite their potential, therapeutic cancer vaccines face challenges such as overcoming the immunosuppressive tumor microenvironment (TME) and ensuring robust immune responses against diverse tumor antigens. Continued research is essential for optimizing vaccine design and improving patient outcomes in cancer therapy.
2.2. Challenges of Predefined Antigen Vaccines Therapeutic Applications
2.2.1. Tumor Endogenous Drug Resistance
Endogenous drug resistance in tumors refers to the inherent ability of cancer cells to resist the effects of therapeutic agents before any treatment is administered. This type of resistance can arise from various factors, including pre-existing genetic mutations, tumor heterogeneity, and the activation of cellular defense mechanisms [25]. For instance, mutations in genes such as KRAS can lead to the production of neoantigens, which may not be effectively targeted by existing vaccines, thus allowing tumors to evade immune detection [26]. The exhaustion of tumor antigens represents a potential immune evasion mechanism, particularly when the depletion of certain antigens does not affect the survival of tumor cells [27]. Additionally, tumor heterogeneity plays a crucial role in resistance mechanisms. Different subpopulations of cancer cells within a single tumor may express varying levels of TAAs or exhibit different immune responses, complicating the effectiveness of vaccines designed to elicit a uniform immune response [28]. Another significant factor contributing to endogenous drug resistance is the activation of intracellular signaling pathways that enhance cell survival and promote DNA repair mechanisms. For example, the activation of the WNT/β-catenin pathway has been linked to reduced immune cell infiltration and poor responses to immune checkpoint blockade therapies [29].
2.2.2. Tumor Exogenous Drug Resistance
The tumor microenvironment (TME) can contribute to exogenous drug resistance by creating an immunosuppressive milieu that inhibits effective immune responses. The presence of cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) can alter the extracellular matrix and secrete various cytokines that enhance tumor cell survival and promote resistance to therapies [30]. In terms of specific examples related to cancer vaccines, research indicates that while therapeutic vaccines can stimulate immune responses against tumors, their effectiveness can be significantly compromised by the immunosuppressive factors present in the TME. For instance, studies on neoantigen vaccines have shown effectiveness in eliciting strong T-cell responses; however, their success is often limited by the presence of regulatory T cells (Tregs) and other immunosuppressive elements within the TME [31]. Factors such as hypoxia, acidity, and the presence of regulatory T cells can suppress T-cell activation and function, diminishing the overall efficacy of a cancer vaccine. For example, studies have shown that tumors often produce immunosuppressive cytokines that can inhibit T-cell proliferation and activity, thereby allowing cancer cells to escape immune surveillance [32]. Moreover, a study discusses how certain neoantigen vaccines can induce immunosuppressive Tr1 cells, which are distinct from traditional Tregs but also inhibit tumor elimination [33]. This highlights the complexity of immune responses elicited by neoantigen vaccines.
2.2.3. Limitations of Predefined Antigen Vaccines and Technical Challenges in Research and Development
Neoantigen vaccines can barely eliminate malignant tumors, partly because some selected neoantigens, for example, melanocyte differentiation antigens, cannot induce continuous and effective tumor regression [34]. When the target neoantigen is a mutated subclone of a gene, a tumor vaccine can only kill a few of the tumor cells, which restricts its clinical efficacy. Personalized neoantigens can elicit tumor-specific T-cell responses; however, once the tumor cells expressing these specific neoantigens are eliminated, tumor cells that do not express the neoantigens would continue their expansion [12]. Though some results of neoantigen vaccination have brought hope to cancer treatment, experiments detecting tumor-specific cells in tumor and peripheral blood T-cell populations indicate that only a small fraction of the predicted useful neoantigens possess spontaneous and durable immunogenicity [35,36]. Therefore, our understanding of epitopes that elicit immunogenic responses remains inadequate, and there are still many obstacles in the selection of neoantigens.
Firstly, the types and quantities of neoantigens observed in each tumor sample are currently limited by sequencing and bioinformatics technologies. Traditional methods may miss more than half of the tumor neoantigens due to potential defects in tumor DNA repair and RNA splicing [37]. Secondly, determining which neoantigens can be presented on MHC molecules and whether they possess immunogenicity remains an unresolved issue. In trials involving multiple neoantigen-targeting vaccines, only 15–30% of the predicted epitopes were able to generate CD8+ T-cell responses, and these responses were relatively weak [38]. Moreover, some tumor vaccines constructed from antigens with low MHC I binding scores have been observed to induce effective antitumor effects, highlighting the shortcomings of current algorithms [39]. Consequently, therapeutic cancer vaccines aimed at treating existing diseases face challenges that are starkly different from those encountered by preventive vaccines targeting infectious pathogens [40].
3. Anonymous Antigen Vaccines
3.1. Anonymous Antigen Vaccines Ex Vivo
The development of anonymous antigen vaccine ex vivo starts from isolating the dendritic cells (DCs) of patients, which are then stimulated with specific antigens in vitro (ex vivo) before being reinfused into the patient. The use of autologous DCs allows for a personalized approach to vaccination, as the therapy is tailored to the individual’s unique tumor antigen profile [41,42,43]. This customization aims to enhance immunogenicity and therapeutic efficacy. By capturing a wide range of tumor antigens, anonymous antigen vaccines ex vivo can potentially elicit a more comprehensive immune response compared to vaccines targeting predefined antigens [44]. This characteristic is particularly advantageous in addressing tumors. Kandalaft et al. [41] conducted a Phase I clinical trial evaluating a dendritic cell vaccine pulsed with autologous oxidized lysate in patients with recurrent ovarian cancer, in which their results demonstrated that this approach was well-tolerated and capable for inducing specific immune responses, with a subset of patients exhibiting prolonged progression-free survival. This study provides foundational evidence for the safety and immunogenicity of dendritic cell-based therapies in ovarian cancer. Chiang et al. [44] further advanced this field by reporting a dendritic cell vaccine pulsed with hypochlorous acid-oxidized ovarian cancer lysate. Their research revealed that this vaccine could elicit broad antitumor immunity in preclinical models and early clinical trials, effectively priming T-cell responses against multiple tumor-associated antigens. This work highlights the importance of optimizing antigen presentation to enhance the efficacy of dendritic cell vaccines. Dhandapani et al. [43] explored the synergistic effects of autologous cervical tumor lysate-pulsed dendritic cell stimulation combined with cisplatin treatment. Their results indicated that this combination therapy could significantly reduce the population of regulatory T cells in vitro, thereby enhancing antitumor immunity and suggesting a potential strategy for overcoming immune suppression in the tumor microenvironment. Nakazawa et al. [42] investigated the capacity of human dendritic cells pulsed with autologous induced pluripotent stem cell (iPSC) lysate to induce cytotoxic T lymphocytes (CTLs) against HLA-A33-matched cancer cells. This study demonstrated effective CTL activation, underscoring the potential for utilizing iPSC-derived antigens to personalize immunotherapeutic strategies. These studies highlight the potential efficacy of dendritic cell vaccines in enhancing immune responses against tumors. Overall, compared to predefined antigen vaccines, anonymous antigen vaccines ex vivo could provide a comprehensive spectrum of tumor antigens and demonstrate significant efficacy in inducing systemic tumor regression [45].
3.2. Anonymous Antigen Vaccines In Situ
The development of tumor vaccines has been limited due to the lack of universal tumor-associated antigens (TAAs) that could activate tumor-specific TILs against the heterogeneity and different kinds of tumor, along with the challenges associated with isolating and preparing personalized vaccines. Therefore, in situ vaccination methods, which aim to initiate or enhance immune responses at the tumor site by utilizing the abundant potential tumor-associated antigens present locally, have garnered significant interest. This approach is referred to as in situ vaccination (ISV) [46]. There are various types of anonymous antigen vaccines in situ, including viruses, pattern recognition receptor (PRR) agonists, other immune stimulators, oncolytic viruses, and oncolytic peptides [47,48,49,50]. These vaccines must effectively induce the recruitment of antigen-presenting cells (APCs), facilitate tumor antigen loading, and activate these cells to successfully initiate the cross-priming of tumor-reactive T cells, thereby inducing systemic antitumor immune responses or vaccine effects [19]. In situ vaccination utilizes the full spectrum of tumor antigens to elicit comprehensive antitumor immunity. Representative results from clinical trial reports are summarized in Table 1.

Table 1.
Anonymous Antigen Vaccines In Situ Entering Clinical Trials.
3.2.1. Flt3L
Flt3L is a dendritic cell (DC) growth factor that can induce rapid DC expansion in vivo. Signals mediated by Flt3L through its receptor Flt3 are essential for the survival, proliferation, and differentiation of DC progenitors and immature DCs. Flt3L promotes the maturation and activation of DCs, enhances the expression of the co-stimulatory molecules and cytokines that facilitate T-cell activation and differentiation, and promotes the migration of DCs to lymphoid organs and other tissues, thereby presenting antigens to T cells and triggering immune responses. Consequently, Flt3L has been utilized as a tool to increase DC numbers in tumor immunotherapy, potentially enhancing the efficacy of such treatments [49,61]. Clinical trials have validated the antitumor efficacy of Flt3L, in which a phase I clinical study reported that injection of an adenoviral vector expressing herpes simplex virus thymidine kinase (HSV-TK) and Flt3L into the tumor cavity of resectable malignant gliomas induced effective antitumor immune responses, resulting in the generation of antitumor memory and new antigen recognition capabilities. Preliminary results indicate that combination therapy with the viral vector improved survival rates, although further research and analysis are required. This clinical trial has received approval from the FDA and multiple institutions and is currently ongoing (NCT01811992) [62]. Overall, these studies suggest that Flt3L may be an effective tool for in situ tumor vaccination, as it can enhance immune responses to tumor antigens and increase the numbers of DCs and other antigen-presenting cells. However, further research is needed to determine the optimal dosing regimen and timing for Flt3L administration, as well as its efficacy in large-scale clinical trials.
3.2.2. TLR Agonists
Toll-like receptors (TLRs) are a class of evolutionarily conserved pattern recognition receptors (PRRs) that play a crucial role in cancer immunotherapy [63]. TLR ligands possess potent antigen-presenting cell (APC) activation capabilities, thereby promoting the induction of antitumor immune responses. They have been utilized as adjuvants in conventional vaccines to enhance the efficacy of tumor immunotherapy [64].
TLR9 is an endosomal receptor that is highly expressed in various mouse DC subsets and predominantly expressed in human B cells and plasmacytoid DCs, but not in conventional DC1 cells that have antigen cross-presentation capabilities. Most TLR9 agonists are unmethylated, low-methylation CpG oligodeoxynucleotides, which can be classified into the categories CpG-A, CpG-B, or CpG-C. These agonists induce the activation of plasmacytoid DCs and B cells, leading to the production of pro-inflammatory cytokines such as type I interferon (IFN). However, a large phase III trial reported an objective response rate (ORR) of 9% for the combination of CpG-B tilsotolimod with ipilimumab in treating refractory melanoma, which is similar to ipilimumab alone. In a trial involving the injection of virus-like particles containing CpG-A (CMP-001) in patients with anti-PD-1 refractory melanoma, monotherapy led to systemic tumor regression, while the combination with pembrolizumab yielded an ORR of 28%. These trials reflect a common issue that while TLR9 agonists can induce intratumoral inflammatory responses, their efficacy remains suboptimal, potentially due to insufficient presentation of tumor antigens. Tumor antigens are primarily presented by cDC1 to CD8+ T cells; however, cDC1 does not express TLR9 and therefore cannot be activated by CpG-A.
TLR3 is primarily expressed on cDC1 and recognizes double-stranded RNA [65]. Poly-ICLC, a complex of polyinosinic-polycytidylic acid, poly-L-lysine, and carboxymethylcellulose, is a widely studied TLR3 agonist that activates different APC subsets through TLR3 and RIG-I-like receptors (RLRs), such as MDA-5. Several peptide-based brain cancer vaccine studies suggest that Poly-ICLC applied in situ may produce positive therapeutic effects [66,67,68]. Additionally, intratumoral injection of Poly-IC significantly inhibited tumor growth and extended survival in mice with melanoma; when combined with tumor-specific T cells or CD40L-expressing plasmids, complete tumor elimination was achieved [69].
The primary ligand for TLR4 is bacterial lipopolysaccharide (LPS), which activates DCs upon binding to TLR4, enhancing antigen presentation and producing various pro-inflammatory cytokines [70,71]. In rodent tumor models, intratumoral injection of LPS has demonstrated antitumor effects without significant toxicity. In subcutaneous glioblastoma mouse models or subcutaneous glioma rat models, intratumoral injection of LPS led to partial and complete tumor regression. Notably, nude mice exhibited lower levels of tumor regression, indicating that T cells play a crucial role in the tumor regression process [72,73]. Another TLR4 agonist (OK-432/Picibanil) used in combination with intratumoral DC injection therapy for pancreatic cancer is currently undergoing clinical trials (NCT00795977). Similarly, a recent study has demonstrated that in situ vaccines can elicit robust immune responses. For instance, the use of cowpea mosaic virus (CPMV) as an in situ vaccine has shown promising results in mouse models, inducing systemic antitumor immunity and triggering regression in both treated and untreated tumors. This strategy also enhances responses to anti-PD-1 therapy, suggesting a synergistic effect when combined with checkpoint inhibitors [74].
3.2.3. Intratumorally Administered Oncolytic Viruses, Oncolytic Bacteria and Oncolytic Peptides
Oncolytic viruses (OVs) is regarded as a promising new class of tumor immunotherapy, which utilize the inherent ability of certain replicative viruses to preferentially infect and lyse tumor cells, demonstrating potential systemic vaccine effects following intratumoral administration [15]. The main advantage of oncolytic viruses is their ability to be engineered to express specific genes, enhancing cross-presentation of tumor antigens, improving the maturation of antigen-presenting cells (APCs), and reducing immune suppression within the tumor microenvironment [46]. Currently, the only oncolytic virus approved by the FDA is Talimogene laherparepvec (TVEC), a genetically modified type 1 herpes simplex virus characterized by reduced virulence and selective replication within tumors. A phase III randomized trial comparing TVEC with GM-CSF demonstrated a significant increase in the objective response rate (ORR) for TVEC [47]. Additionally, a multicenter phase II study indicated that TVEC could induce the regression of non-injected lesions [75]. Despite many strategies showing antitumor efficacy in mouse models and various cancer types, response rates remain highly variable [46].
Similarly to oncolytic viruses, research has explored the use of oncolytic bacteria for tumor immunotherapy. For instance, researchers developed a photosensitizer-labeled anaerobic oncolytic bacterium based on metabolic engineering. After injection into melanoma tumors, these anaerobic bacteria proliferate in hypoxic regions of the tumor, competing for nutrients and thereby limiting tumor growth and causing the lysis of the tumor cells. The photosensitizer on the bacteria exerts photodynamic effects in oxygen-rich areas to further eliminate residual melanoma. This engineered oncolytic bacterium effectively eradicated melanoma in mice and exhibited ideal biocompatibility, presenting a promising approach for eradicating solid tumors [76].
Furthermore, increasing evidence supports the value of oncolytic peptides as anticancer agents. Despite intra-tumor heterogeneity (ITH), oncolytic peptides can induce robust antitumor immune responses by promoting the infiltration of cytotoxic T lymphocytes (CTLs) and other immune effector cells into tumors while preferentially killing malignant cells [50]. Some oncolytic peptides can trigger immunogenic cell death (ICD), activating and recruiting immune effector cells [77]. In a phase I dose-escalation study involving intratumoral administration in patients with advanced solid tumors, the oncolytic peptide LTX-315 demonstrated ideal patient tolerance and therapeutic efficacy, including tumor regression in some patients and a significant reduction in tumor volume in 30% of cases, promoting CD8+ T-cell infiltration within tumors and inducing distant responses [59]. However, clinical development of these agents for tumor treatment remains in its early stages [50].
4. Advantages of Anonymous Antigen Vaccines In Situ
In situ vaccination (ISV) represents a method of forming tumor vaccines in vivo, which significantly reduces the technical obstacles in ex vivo vaccine development, thereby facilitating faster vaccine development. Furthermore, this strategy is not limited to single tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs), but rather can utilize all the tumor antigens in a patient. To elicit a robust memory antitumor immune response, an in situ vaccine should be capable of inducing immunogenic cell death (ICD) in tumors, promoting the release of TAAs and enhancing antigen-presenting cell (APC) uptake and activation, which in turn induces T-cell responses and activates systemic antitumor immunity. Since fully activated effector T cells can kill tumor cells independently of co-stimulatory signals and are less sensitive to inhibitory signals, potent antitumor T cells generated at one tumor site can also attack distant tumor lesions. Therefore, an effective in situ vaccination strategy requires the treatment of a single tumor site to induce the release of tumor antigens and the occurrence of ICD, leading to a systemic immune response and tumor regression, with this delivery site being personalized for each patient [40]. As immunotherapy increasingly becomes a core component of cancer treatment, there is a growing demand for methods to enhance the responsiveness to immune checkpoint blockade (ICB) therapies [40]. Anonymous antigen vaccines in situ are likely to be the most promising approaches to achieve this goal by transforming patients from low-T-cell infiltration to a high-T-cell infiltration phenotype, thereby improving the responsiveness to ICB therapies [78,79].
5. Conclusions
The primary objective of cancer immunotherapy is to induce a strong antitumor immune response without affecting normal cells in the body. The encouraging efficacy demonstrated by predefined antigen vaccines has opened new avenues for cancer immunotherapy; however, techniques to predict, identify, and screen for the correct antigen epitopes remain insufficient. Anonymous antigen vaccines in situ can leverage the complete tumor antigen repertoire from cancer patients, providing a clearer individual tumor profile that current technologies could not offer while minimizing the risk of immune escape due to tumor variability. The next generation of in situ vaccines has shown considerable efficacy as a new direction in tumor vaccine therapy, and further evaluation of the immune effects and clinical efficacy induced by various in situ vaccination strategies across different tumor types is still needed.
Author Contributions
Conceptualization, D.P., J.L. and X.H.; validation, L.Y. and Y.Z.Z.; investigation, D.P., J.L., K.K. and Y.Y.; resources, L.Y.; data curation, L.Y.; writing—original draft preparation, D.P., J.L. and S.W.; writing—review and editing, D.P., J.L., X.H., S.W., K.K., Y.Y., Y.Z.Z. and L.Y.; supervision, L.Y. and Y.Z.Z.; funding acquisition, L.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Macau Science and Technology Development Fund, grant number [FDCT 0148/2022/A3 and 0019/2024/RIA1], and the APC was funded by [FDCT 0148/2022/A3].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data relevant to the study are included in the article. Additional data are available upon reasonable request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mukherjee, A.G.; Wanjari, U.R.; Namachivayam, A.; Murali, R.; Prabakaran, D.S.; Ganesan, R.; Renu, K.; Dey, A.; Vellingiri, B.; Ramanathan, G.; et al. Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach. Vaccines 2022, 10, 1493. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Von Rueden, S.K.; Fan, T.M. Cancer-Immunity Cycle and Therapeutic Interventions- Opportunities for Including Pet Dogs with Cancer. Front. Oncol. 2021, 11, 773420. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Zhao, X.; Subramanian, S. Intrinsic Resistance of Solid Tumors to Immune Checkpoint Blockade Therapy. Cancer Res. 2017, 77, 817–822. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, Q.; Zhao, X.; Zhao, R.; Wang, Y.; Wang, Y.; Liu, J.; Shang, Y.; Zhao, S.; Wu, T.; et al. A DNA nanodevice-based vaccine for cancer immunotherapy. Nat. Mater. 2021, 20, 421–430. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, G.; Tang, T.Y.; Gao, X.; Liang, T.B. Personalized pancreatic cancer therapy: From the perspective of mRNA vaccine. Mil. Med. Res. 2022, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.C.; Castiello, L.; Mattei, M.; Santodonato, L.; D’Agostino, G.; Muraro, E.; Martorelli, D.; Lapenta, C.; Di Napoli, A.; Di Landro, F.; et al. Clinical and Antitumor Immune Responses in Relapsed/Refractory Follicular Lymphoma Patients after Intranodal Injections of IFNα-Dendritic Cells and Rituximab: A Phase I Clinical Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5231–5241. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef]
- Tiptiri-Kourpeti, A.; Spyridopoulou, K.; Pappa, A.; Chlichlia, K. DNA vaccines to attack cancer: Strategies for improving immunogenicity and efficacy. Pharmacol. Ther. 2016, 165, 32–49. [Google Scholar] [CrossRef]
- Jou, J.; Harrington, K.J.; Zocca, M.B.; Ehrnrooth, E.; Cohen, E.E.W. The Changing Landscape of Therapeutic Cancer Vaccines-Novel Platforms and Neoantigen Identification. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Qi, X.W.; Zhang, F.; Wu, H.; Liu, J.L.; Zong, B.G.; Xu, C.; Jiang, J. Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 8924. [Google Scholar] [CrossRef]
- Katanasaka, Y.; Kodera, Y.; Kitamura, Y.; Morimoto, T.; Tamura, T.; Koizumi, F. Epidermal growth factor receptor variant type III markedly accelerates angiogenesis and tumor growth via inducing c-myc mediated angiopoietin-like 4 expression in malignant glioma. Mol. Cancer 2013, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Taberna, M.; Mena, M.; Pavón, M.A.; Alemany, L.; Gillison, M.L.; Mesía, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.E.; Gottlieb, A.P.; Park, K.S.; Zhao, Z.; Mitragotri, S. Cancer vaccines in the clinic. Bioeng. Transl. Med. 2024, 9, e10588. [Google Scholar] [CrossRef] [PubMed]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef]
- Zhou, C.; Li, W.; Liang, Z.; Wu, X.; Cheng, S.; Peng, J.; Zeng, K.; Li, W.; Lan, P.; Yang, X.; et al. Mutant KRAS-activated circATXN7 fosters tumor immunoescape by sensitizing tumor-specific T cells to activation-induced cell death. Nat. Commun. 2024, 15, 499. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Lin, Z.; Meng, X.; Wen, J.; Corral, J.M.; Andreev, D.; Kachler, K.; Schett, G.; Chen, X.; Bozec, A. Intratumor Heterogeneity Correlates with Reduced Immune Activity and Worse Survival in Melanoma Patients. Front. Oncol. 2020, 10, 596493. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Takeuchi, Y.; Ward, J.P.; Sharma, N.; Liu, T.-T.; Sukhov, V.; Firulyova, M.; Song, Y.; Ameh, S.; Brioschi, S.; et al. Neoantigen-specific cytotoxic Tr1 CD4 T cells suppress cancer immunotherapy. Nature 2024, 632, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef]
- Lam, H.; McNeil, L.K.; Starobinets, H.; DeVault, V.L.; Cohen, R.B.; Twardowski, P.; Johnson, M.L.; Gillison, M.L.; Stein, M.N.; Vaishampayan, U.N.; et al. An Empirical Antigen Selection Method Identifies Neoantigens That Either Elicit Broad Antitumor T-cell Responses or Drive Tumor Growth. Cancer Discov. 2021, 11, 696–713. [Google Scholar] [CrossRef]
- Wu, Y.M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e1714. [Google Scholar] [CrossRef]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Ebrahimi-Nik, H.; Moussa, M.; Englander, R.P.; Singhaviranon, S.; Michaux, J.; Pak, H.; Miyadera, H.; Corwin, W.L.; Keller, G.L.J.; Hagymasi, A.T.; et al. Reversion analysis reveals the in vivo immunogenicity of a poorly MHC I-binding cancer neoepitope. Nat. Commun. 2021, 12, 6423. [Google Scholar] [CrossRef]
- Sellars, M.C.; Wu, C.J.; Fritsch, E.F. Cancer vaccines: Building a bridge over troubled waters. Cell 2022, 185, 2770–2788. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Chiang, C.L.; Tanyi, J.; Motz, G.; Balint, K.; Mick, R.; Coukos, G. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Maeoka, R.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Nishimura, F.; Yamada, S.; Nakagawa, I.; Park, Y.S.; Nakase, H.; et al. Capability of Human Dendritic Cells Pulsed with Autologous Induced Pluripotent Stem Cell Lysate to Induce Cytotoxic T Lymphocytes against HLA-A33-Matched Cancer Cells. Int. J. Mol. Sci. 2022, 23, 12992. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, H.; Seetharaman, A.; Jayakumar, H.; Ganeshrajah, S.; Singh, S.S.; Thangarajan, R.; Ramanathan, P. Autologous cervical tumor lysate pulsed dendritic cell stimulation followed by cisplatin treatment abrogates FOXP3+ cells in vitro. J. Gynecol. Oncol. 2021, 32, e59. [Google Scholar] [CrossRef]
- Chiang, C.L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Zappasodi, R.; Carlo-Stella, C.; Mortarini, R.; Pupa, S.M.; Magni, M.; Devizzi, L.; Matteucci, P.; Baldassari, P.; Ravagnani, F.; et al. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: A pilot study. Blood 2009, 113, 18–27. [Google Scholar] [CrossRef]
- Hammerich, L.; Bhardwaj, N.; Kohrt, H.E.; Brody, J.D. In situ vaccination for the treatment of cancer. Immunotherapy 2016, 8, 315–330. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Márquez-Rodas, I.; Longo, F.; Rodriguez-Ruiz, M.E.; Calles, A.; Ponce, S.; Jove, M.; Rubio-Viqueira, B.; Perez-Gracia, J.L.; Gómez-Rueda, A.; López-Tarruella, S.; et al. Intratumoral nanoplexed poly I:C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci. Transl. Med. 2020, 12, eabb0391. [Google Scholar] [CrossRef]
- Cueto, F.J.; Sancho, D. The Flt3L/Flt3 Axis in Dendritic Cell Biology and Cancer Immunotherapy. Cancers 2021, 13, 1525. [Google Scholar] [CrossRef]
- Vitale, I.; Yamazaki, T.; Wennerberg, E.; Sveinbjørnsson, B.; Rekdal, Ø.; Demaria, S.; Galluzzi, L. Targeting Cancer Heterogeneity with Immune Responses Driven by Oncolytic Peptides. Trends Cancer 2021, 7, 557–572. [Google Scholar] [CrossRef]
- NCT01811992. Combined Cytotoxic and Immune-Stimulatory Therapy for Glioma. ClinicalTrials.gov Database 2021. Available online: https://clinicaltrials.gov/study/NCT01811992 (accessed on 14 November 2024).
- NCT01976585. In Situ Vaccine for Low-Grade Lymphoma: Combination of Intratumoral Flt3L and Poly-ICLC with Low-Dose Radiotherapy. ClinicalTrials.gov Database 2020. Available online: https://clinicaltrials.gov/study/NCT01976585 (accessed on 14 November 2024).
- NCT03729245. A Study of Bempegaldesleukin (NKTR-214: BEMPEG) in Combination with Nivolumab Compared with the Investigator’s Choice of a Tyrosine Kinase Inhibitor (TKI) Therapy (Either Sunitinib or Cabozantinib Monotherapy) for Advanced Metastatic Renal Cell Carcinoma (RCC). ClinicalTrials.gov Database 2022. Available online: https://clinicaltrials.gov/study/NCT03729245 (accessed on 14 November 2024).
- NCT03445533. A Study of Tilsotolimod in Combo with Ipilimumab vs Ipilimumab Alone in Subjects with Anti-PD-1 Refractory Melanoma. ClinicalTrials.gov Database 2021. Available online: https://clinicaltrials.gov/study/NCT03445533 (accessed on 14 November 2024).
- El Haddaoui, H.; Brood, R.; Latifi, D.; Oostvogels, A.A.; Klaver, Y.; Moskie, M.; Mustafa, D.A.; Debets, R.; van Eijck, C.H.J. Rintatolimod (Ampligen®) Enhances Numbers of Peripheral B Cells and Is Associated with Longer Survival in Patients with Locally Advanced and Metastasized Pancreatic Cancer Pre-Treated with FOLFIRINOX: A Single-Center Named Patient Program. Cancers 2022, 14, 1377. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Saito, T.; Kenjo, A.; Hoshino, M.; Terashima, M.; Sato, T.; Anazawa, T.; Kimura, T.; Tsuchiya, T.; Irisawa, A.; et al. Phase I trial of preoperative intratumoral injection of immature dendritic cells and OK-432 for resectable pancreatic cancer patients. J. Hepatobiliary Pancreat. Sci. 2012, 19, 465–475. [Google Scholar] [CrossRef] [PubMed]
- NCT02180698. TLR4 Agonist GLA-SE and Radiation Therapy in Treating Patients with Soft Tissue Sarcoma That Is Metastatic or Cannot Be Removed by Surgery. ClinicalTrials.gov Database 2016. Available online: https://clinicaltrials.gov/study/NCT02180698 (accessed on 14 November 2024).
- NCT02773849. ADSTILADRIN (=INSTILADRIN) in Patients with High-Grade, Bacillus Calmette-Guerin (BCG) Unresponsive Non-Muscle Invasive Bladder Cancer (NMIBC). ClinicalTrials.gov Database 2023. Available online: https://clinicaltrials.gov/study/NCT02773849 (accessed on 14 November 2024).
- Spicer, J.; Marabelle, A.; Baurain, J.F.; Jebsen, N.L.; Jøssang, D.E.; Awada, A.; Kristeleit, R.; Loirat, D.; Lazaridis, G.; Jungels, C.; et al. Safety, Antitumor Activity, and T-cell Responses in a Dose-Ranging Phase I Trial of the Oncolytic Peptide LTX-315 in Patients with Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2755–2763. [Google Scholar] [CrossRef]
- NCT02045602. Phase I Dose Escalation Study of Intravenous VCN-01 with or Without Gemcitabine and Abraxane® in Patients with Advanced Solid Tumors. ClinicalTrials.gov Database 2020. Available online: https://clinicaltrials.gov/study/NCT02045602 (accessed on 14 November 2024).
- Cheng, H.; Chen, W.; Lin, Y.; Zhang, J.; Song, X.; Zhang, D. Signaling pathways involved in the biological functions of dendritic cells and their implications for disease treatment. Mol. Biomed. 2023, 4, 15. [Google Scholar] [CrossRef]
- Lowenstein, P.R.; Orringer, D.A.; Sagher, O.; Heth, J.; Hervey-Jumper, S.L.; Mammoser, A.G.; Junck, L.; Leung, D.; Umemura, Y.; Lawrence, T.S.; et al. First-in-human phase I trial of the combination of two adenoviral vectors expressing HSV1-TK and FLT3L for the treatment of newly diagnosed resectable malignant glioma: Initial results from the therapeutic reprogramming of the brain immune system. J. Clin. Oncol. 2019, 37, 2019. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. BioMed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Zhu, X.; Nishimura, F.; Sasaki, K.; Fujita, M.; Dusak, J.E.; Eguchi, J.; Fellows-Mayle, W.; Storkus, W.J.; Walker, P.R.; Salazar, A.M.; et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J. Transl. Med. 2007, 5, 10. [Google Scholar] [CrossRef]
- Zhu, X.; Fallert-Junecko, B.A.; Fujita, M.; Ueda, R.; Kohanbash, G.; Kastenhuber, E.R.; McDonald, H.A.; Liu, Y.; Kalinski, P.; Reinhart, T.A.; et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol. Immunother. 2010, 59, 1401–1409. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Chamberlain, M.C.; Grossman, S.A.; Peereboom, D.M.; Lesser, G.J.; Batchelor, T.T.; Desideri, S.; Salazar, A.M.; Ye, X. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 2010, 12, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.M.; Pegram, H.J.; Westwood, J.A.; John, L.B.; Devaud, C.; Clarke, C.J.; Restifo, N.P.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Adoptive immunotherapy combined with intratumoral TLR agonist delivery eradicates established melanoma in mice. Cancer Immunol. Immunother. 2011, 60, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 2006, 440, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, M.R.; Won, E.K.; Zahner, M.C. Intratumoral injection of lipopolysaccharide causes regression of subcutaneously implanted mouse glioblastoma multiforme. Neurosurgery 2001, 48, 607–614, discussion 614–605. [Google Scholar] [CrossRef]
- Mariani, C.L.; Rajon, D.; Bova, F.J.; Streit, W.J. Nonspecific immunotherapy with intratumoral lipopolysaccharide and zymosan A but not GM-CSF leads to an effective anti-tumor response in subcutaneous RG-2 gliomas. J. Neurooncol 2007, 85, 231–240. [Google Scholar] [CrossRef]
- Mao, C.; Beiss, V.; Ho, G.W.; Fields, J.; Steinmetz, N.F.; Fiering, S. In situ vaccination with cowpea mosaic virus elicits systemic antitumor immunity and potentiates immune checkpoint blockade. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Amatruda, T.; Reid, T.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; Nemunaitis, J.; Zloza, A.; Wolf, M.; et al. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J. Immunother. Cancer 2016, 4, 12. [Google Scholar] [CrossRef]
- Shi, L.; Liu, X.; Li, Y.; Li, S.; Wu, W.; Gao, X.; Liu, B. Living Bacteria-Based Immuno-Photodynamic Therapy: Metabolic Labeling of Clostridium butyricum for Eradicating Malignant Melanoma. Adv. Sci. 2022, 9, e2105807. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).