
Immunogenicity of Tetravalent Protein Vaccine SCTV01E-2 against SARS-CoV-2 EG.5 Subvaraint: A Phase 2 Trial
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Randomization and Masking
2.3. Procedures
2.4. Outcomes
2.5. Statistical Analysis
3. Results
3.1. Demographic and Baseline Characteristics
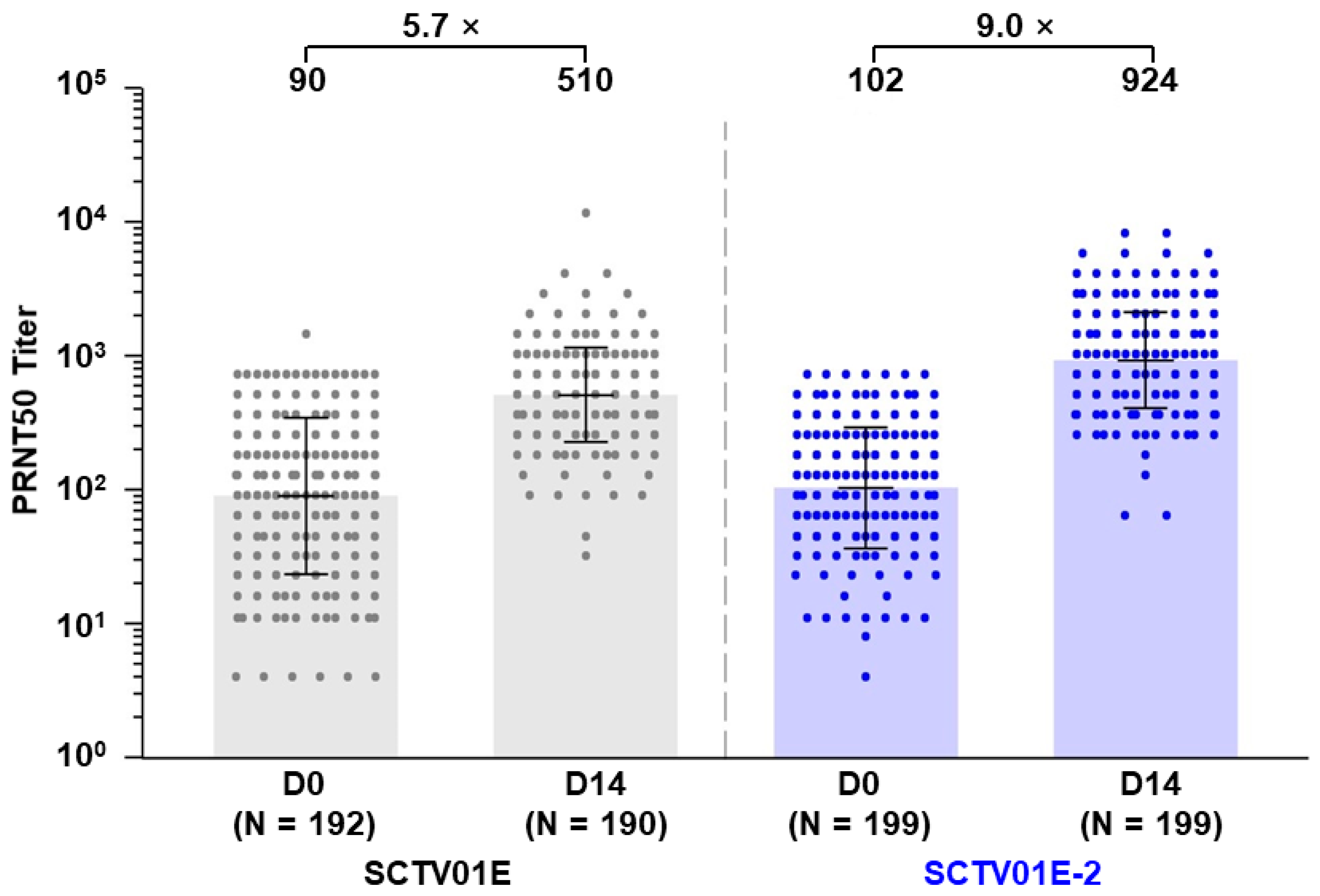
3.2. Geometric Mean Titer (GMT) and Seroconversion Rate (SRR) of Neutralizing Antibodies against Omicron EG.5
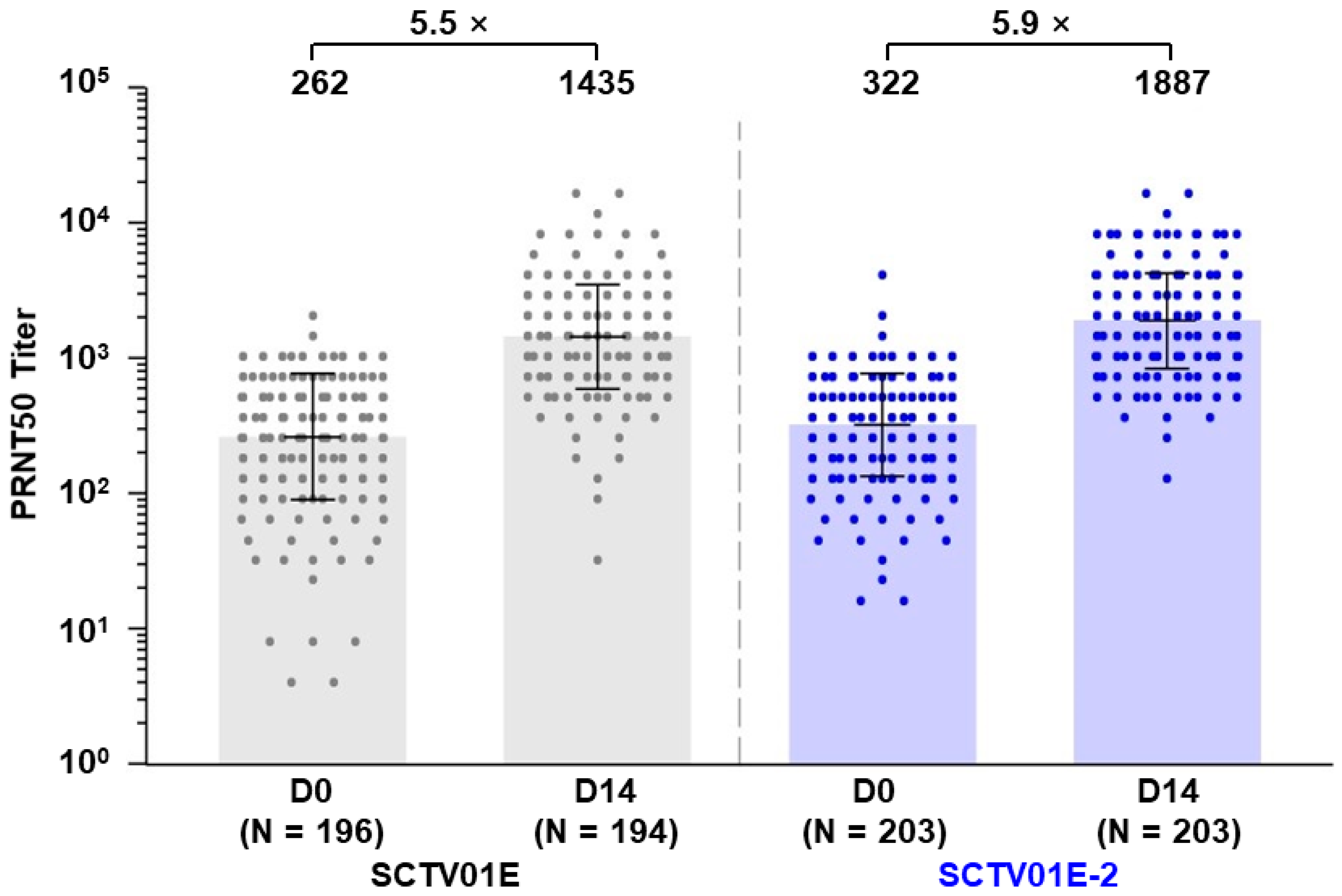
3.3. Geometric Mean Titer (GMT) and Seroconversion Rate (SRR) of Neutralizing Antibodies against Omicron XBB.1
3.4. Subgroup Analyses of nAb Responses to Omicron EG.5 and XBB.1
3.5. Vaccine Immunogenicity and Effectiveness Will Continue to Be Monitored
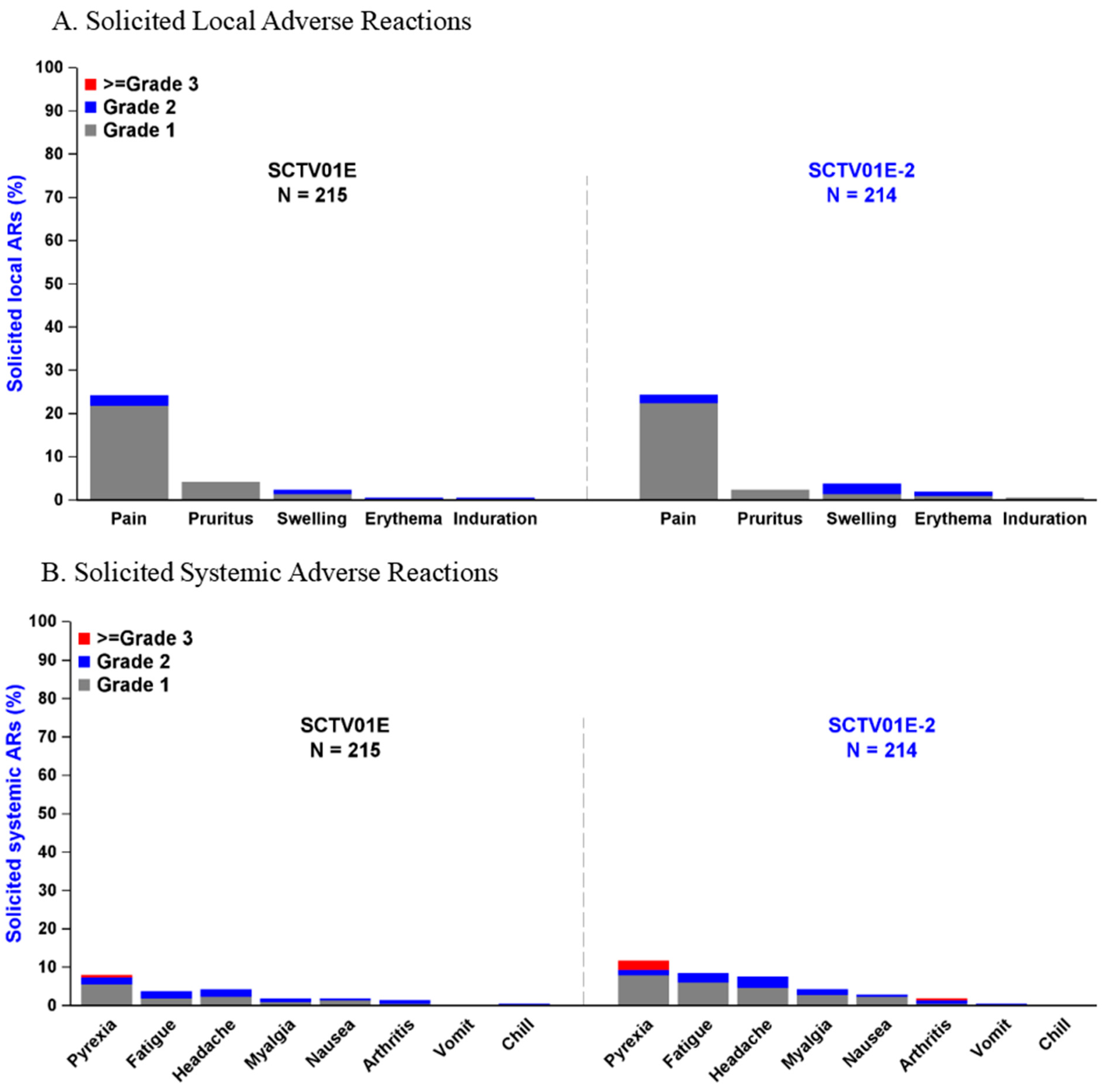
3.6. Adverse Events
3.7. Subgroup Analyses of Adverse Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Hachmann, N.P.; Miller, J.; Collier, A.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K.; et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef]
- Parums, D.V. Editorial: A Rapid Global Increase in COVID-19 is Due to the Emergence of the EG.5 (Eris) Subvariant of Omicron SARS-CoV-2. Med. Sci. Monit. 2023, 29, e942244. [Google Scholar] [CrossRef]
- Abbasi, J. What to Know About EG.5, the Latest SARS-CoV-2 “Variant of Interest”. JAMA 2023, 330, 900–901. [Google Scholar] [CrossRef]
- Dyer, O. COVID-19: Infections climb globally as EG.5 variant gains ground. BMJ 2023, 382, 1900. [Google Scholar] [CrossRef] [PubMed]
- Zappa, M.; Verdecchia, P.; Andolina, A.; Angeli, F. The old and the new: The EG.5 (‘Eris’) sub-variant of Coronavirus. Eur. J. Intern. Med. 2023, 117, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Darvishi, M.; Bezmin Abadi, A.T. ‘The end’—or is it? Emergence of SARS-CoV-2 EG.5 and BA.2.86 subvariants. Future Virol. 2023, 18, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kempf, A.; Nehlmeier, I.; Cossmann, A.; Dopfer-Jablonka, A.; Stankov, M.V.; Schulz, S.R.; Jack, H.M.; Behrens, G.M.N.; Pohlmann, S.; et al. Neutralisation sensitivity of SARS-CoV-2 lineages EG.5.1 and XBB.2.3. Lancet Infect. Dis. 2023, 23, e391–e392. [Google Scholar] [CrossRef] [PubMed]
- Faraone, J.N.; Qu, P.; Goodarzi, N.; Zheng, Y.M.; Carlin, C.; Saif, L.J.; Oltz, E.M.; Xu, K.; Jones, D.; Gumina, R.J.; et al. Immune Evasion and Membrane Fusion of SARS-CoV-2 XBB Subvariants EG.5.1 and XBB.2.3. Emerg. Microbes Infect. 2023, 12, 2270069. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Pascarella, S.; Ciccozzi, A.; Giovanetti, M.; Azzena, I.; Locci, C.; Casu, M.; Fiori, P.L.; Quaranta, M.; Cella, E.; et al. Genetic and structural analyses reveal the low potential of the SARS-CoV-2 EG.5 variant. J. Med. Virol. 2023, 95, e29075. [Google Scholar] [CrossRef] [PubMed]
- EG.5 Initial Risk Evaluation. Available online: https://www.who.int/docs/default-source/coronaviruse/09082023eg.5_ire_final.pdf (accessed on 9 August 2023).
- Wang, Q.; Guo, Y.; Zhang, R.M.; Ho, J.; Mohri, H.; Valdez, R.; Manthei, D.M.; Gordon, A.; Liu, L.; Ho, D.D. Antibody neutralisation of emerging SARS-CoV-2 subvariants: EG.5.1 and XBC.1.6. Lancet Infect. Dis. 2023, 23, e397–e398. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Kosugi, Y.; Uriu, K.; Ito, J.; Hinay, A.A., Jr.; Kuramochi, J.; Sadamasu, K.; Yoshimura, K.; Asakura, H.; Nagashima, M.; et al. Antiviral efficacy of the SARS-CoV-2 XBB breakthrough infection sera against omicron subvariants including EG.5. Lancet Infect. Dis. 2023, 23, e395–e396. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023, 29, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Huang, H.; Yu, C.; Sun, C.; Ma, J.; Kong, D.; Lin, Y.; Zhao, D.; Zhou, S.; Lu, J.; et al. A spike-trimer protein-based tetravalent COVID-19 vaccine elicits enhanced breadth of neutralization against SARS-CoV-2 Omicron subvariants and other variants. Sci. China Life Sci. 2023, 66, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Hannawi, S.; Yan, L.; Saifeldin, L.; Abuquta, A.; Alamadi, A.; Mahmoud, S.A.; Hassan, A.; Zhang, M.; Gao, C.; Chen, Y.; et al. Safety and immunogenicity of multivalent SARS-CoV-2 protein vaccines: A randomized phase 3 trial. EClinicalMedicine 2023, 64, 102195. [Google Scholar] [CrossRef] [PubMed]
- Hannawi, S.; Saifeldin, L.; Abuquta, A.; Alamadi, A.; Mahmoud, S.A.; Hassan, A.; Xu, S.; Li, J.; Liu, D.; Baidoo, A.A.H.; et al. Safety and immunogenicity of a tetravalent and bivalent SARS-CoV-2 protein booster vaccine in men. Nat. Commun. 2023, 14, 4043. [Google Scholar] [CrossRef]
- A Study to Evaluate the Immunogenicity and Safety of a Recombinant Protein COVID-19 Vaccine as Booster Vaccines. Available online: https://clinicaltrials.gov/study/NCT05933512?term=NCT05933512&rank=1 (accessed on 4 July 2023).
- Mahmoud, S.; Ganesan, S.; Al Kaabi, N.; Naik, S.; Elavalli, S.; Gopinath, P.; Ali, A.M.; Bazzi, L.; Warren, K.; Zaher, W.A.; et al. Immune response of booster doses of BBIBP-CORV vaccines against the variants of concern of SARS-CoV-2. J. Clin. Virol. 2022, 150–151, 105161. [Google Scholar] [CrossRef]
- Hannawi, S.; Saifeldin, L.; Abuquta, A.; Alamadi, A.; Mahmoud, S.A.; Hassan, A.; Liu, D.; Yan, L.; Xie, L. Safety and immunogenicity of a bivalent SARS-CoV-2 protein booster vaccine, SCTV01C, in adults previously vaccinated with mRNA vaccine: A randomized, double-blind, placebo-controlled phase 1/2 clinical trial. EBioMedicine 2023, 87, 104386. [Google Scholar] [CrossRef]
- Guidelines for Grading Adverse Events in Preventive Vaccine Clinical Trials. 2019. Available online: https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20191231111901460.html (accessed on 31 December 2019).
- Liu, C.; Zhou, D.; Nutalai, R.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Mentzer, A.J.; Wang, B.; et al. The antibody response to SARS-CoV-2 Beta underscores the antigenic distance to other variants. Cell Host Microbe 2022, 30, 53–68.e12. [Google Scholar] [CrossRef]
- Singh, J.; Malhotra, A.G.; Biswas, D.; Shankar, P.; Lokhande, L.; Yadav, A.K.; Raghuvanshi, A.; Kale, D.; Nema, S.; Saigal, S.; et al. Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India. Genes 2021, 12, 1803. [Google Scholar] [CrossRef]
- Zeng, B.; Gao, L.; Zhou, Q.; Yu, K.; Sun, F. Effectiveness of COVID-19 vaccines against SARS-CoV-2 variants of concern: A systematic review and meta-analysis. BMC Med. 2022, 20, 200. [Google Scholar] [CrossRef]
- Choi, A.; Koch, M.; Wu, K.; Chu, L.; Ma, L.; Hill, A.; Nunna, N.; Huang, W.; Oestreicher, J.; Colpitts, T.; et al. Safety and immunogenicity of SARS-CoV-2 variant mRNA vaccine boosters in healthy adults: An interim analysis. Nat. Med. 2021, 27, 2025–2031. [Google Scholar] [CrossRef]
- Wang, R.; Sun, C.; Ma, J.; Yu, C.; Kong, D.; Chen, M.; Liu, X.; Zhao, D.; Gao, S.; Kou, S.; et al. A Bivalent COVID-19 Vaccine Based on Alpha and Beta Variants Elicits Potent and Broad Immune Responses in Mice against SARS-CoV-2 Variants. Vaccines 2022, 10, 702. [Google Scholar] [CrossRef]
- Beta-Variant Recombinant SARS-CoV-2 Vaccine Induces Durable Cross-Reactive Antibodies against Omicron Variants. Available online: https://www.researchsquare.com/article/rs-2513105/v1 (accessed on 30 January 2023).
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, S.; McGhee, N.; Whatley, J.L.; Essink, B.; Brosz, A.; Tomassini, J.E.; Girard, B.; Wu, K.; Edwards, D.K.; Nasir, A.; et al. Safety and Immunogenicity of XBB.1.5-Containing mRNA Vaccines. MedRxiv 2023. [Google Scholar] [CrossRef]
- COVID-19 Vaccines for 2023–2024. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines-2023-2024 (accessed on 4 October 2023).
- Poon, R.W.; Chan, B.P.; Chan, W.M.; Fong, C.H.; Zhang, X.; Lu, L.; Chen, L.L.; Lam, J.Y.; Cheng, V.C.; Wong, S.S.Y.; et al. SARS-CoV-2 IgG seropositivity after the severe Omicron wave of COVID-19 in Hong Kong. Emerg. Microbes Infect. 2022, 11, 2116–2119. [Google Scholar] [CrossRef]
- Singh, P.; Ujjainiya, R.; Prakash, S.; Naushin, S.; Sardana, V.; Bhatheja, N.; Singh, A.P.; Barman, J.; Kumar, K.; Gayali, S.; et al. A machine learning-based approach to determine infection status in recipients of BBV152 (Covaxin) whole-virion inactivated SARS-CoV-2 vaccine for serological surveys. Comput. Biol. Med. 2022, 146, 105419. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCTV01E (N = 215) n (%) | SCTV01E-2 (N = 214) n (%) | Total (N = 429) n (%) | |
|---|---|---|---|
| Age (Years) | |||
| N | 215 | 214 | 429 |
| Mean (SD) | 56.1 (12.1) | 55.1 (11.5) | 55.6 (11.8) |
| Median (Min, Max) | 58.0 (20, 81) | 58.0 (19, 83) | 58.0 (19, 83) |
| Age subgroups—randomization, n (%) | |||
| 18–59 years | 130 (60.5) | 128 (59.8) | 258 (60.1) |
| ≥60 years | 85 (39.5) | 86 (40.2) | 171 (39.9) |
| Sex, n (%) | |||
| Male | 101 (47.0) | 98 (45.8) | 199 (46.4) |
| Female | 114 (53.0) | 116 (54.2) | 230 (53.6) |
| Nation, n (%) | |||
| Han | 214 (99.5) | 214 (100.0) | 428 (99.8) |
| Others | 1 (0.5) | 0 | 1 (0.2) |
| BMI (kg/m2) ‡ | |||
| N | 215 | 214 | 429 |
| Mean (SD) | 26.2 (3.5) | 26.5 (3.5) | 26.4 (3.5) |
| Median (Min, Max) | 26.0 (18.9, 38.6) | 26.50 (16.4, 39.8) | 26.20 (16.4, 39.8) |
| History of SARS-CoV-2 infection, n (%) | |||
| Yes | 57 (26.5) | 56 (26.2) | 113 (26.3) |
| No | 158 (73.5) | 158 (73.8) | 316 (73.7) |
| Previous vaccination/infection interval, n (%) | |||
| 6–11 months | 67 (31.2) | 67 (31.3) | 134 (31.2) |
| ≥12 months | 148 (68.8) | 147 (68.7) | 295 (68.8) |
| IgM at baseline | |||
| Positive | 17 (7.9) | 11 (5.1) | 28 (6.5) |
| Negative | 198 (92.1) | 203 (94.9) | 401 (93.5) |
| Booster dose of COVID-19 vaccine, n (%) | |||
| Yes | 157 (73.0) | 155 (72.4) | 312 (72.7) |
| No | 58 (27.0) | 59 (27.6) | 117 (27.3) |
| Type of last received COVID-19 vaccine—randomization, n (%) | |||
| Inactive vaccine | 98 (45.6) | 103 (48.1) | 201 (46.9) |
| Adenovirus vector vaccine | 42 (19.5) | 35 (16.4) | 77 (17.9) |
| Recombinant protein vaccine | 75 (34.9) | 76 (35.5) | 151 (35.2) |
| Other vaccines | 0 | 0 | 0 |
| Pre-existing comorbidities, n (%) | |||
| Yes | 84 (39.1) | 98 (45.8) | 182 (42.4) |
| No | 131 (60.9) | 116 (54.2) | 247 (57.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Xu, Q.; Zhu, C.; Xuan, K.; Li, T.; Li, Q.; Pang, X.; Zha, Z.; Li, J.; Qiao, L.; et al. Immunogenicity of Tetravalent Protein Vaccine SCTV01E-2 against SARS-CoV-2 EG.5 Subvaraint: A Phase 2 Trial. Vaccines 2024, 12, 175. https://doi.org/10.3390/vaccines12020175
Tang J, Xu Q, Zhu C, Xuan K, Li T, Li Q, Pang X, Zha Z, Li J, Qiao L, et al. Immunogenicity of Tetravalent Protein Vaccine SCTV01E-2 against SARS-CoV-2 EG.5 Subvaraint: A Phase 2 Trial. Vaccines. 2024; 12(2):175. https://doi.org/10.3390/vaccines12020175
Chicago/Turabian StyleTang, Jihai, Qinghua Xu, Chaoyin Zhu, Kun Xuan, Tao Li, Qingru Li, Xingya Pang, Zhenqiu Zha, Jinwei Li, Liyang Qiao, and et al. 2024. "Immunogenicity of Tetravalent Protein Vaccine SCTV01E-2 against SARS-CoV-2 EG.5 Subvaraint: A Phase 2 Trial" Vaccines 12, no. 2: 175. https://doi.org/10.3390/vaccines12020175
APA StyleTang, J., Xu, Q., Zhu, C., Xuan, K., Li, T., Li, Q., Pang, X., Zha, Z., Li, J., Qiao, L., Xu, H., Wu, G., Tian, Y., Han, J., Gao, C., Yi, J., Qian, G., Tian, X., & Xie, L. (2024). Immunogenicity of Tetravalent Protein Vaccine SCTV01E-2 against SARS-CoV-2 EG.5 Subvaraint: A Phase 2 Trial. Vaccines, 12(2), 175. https://doi.org/10.3390/vaccines12020175