
Polymeric Nanoparticles as Oral and Intranasal Peptide Vaccine Delivery Systems: The Role of Shape and Conjugation

 , , ,
, , ,  ,
,  ,
,  ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Synthesis and Physiochemical Characterization
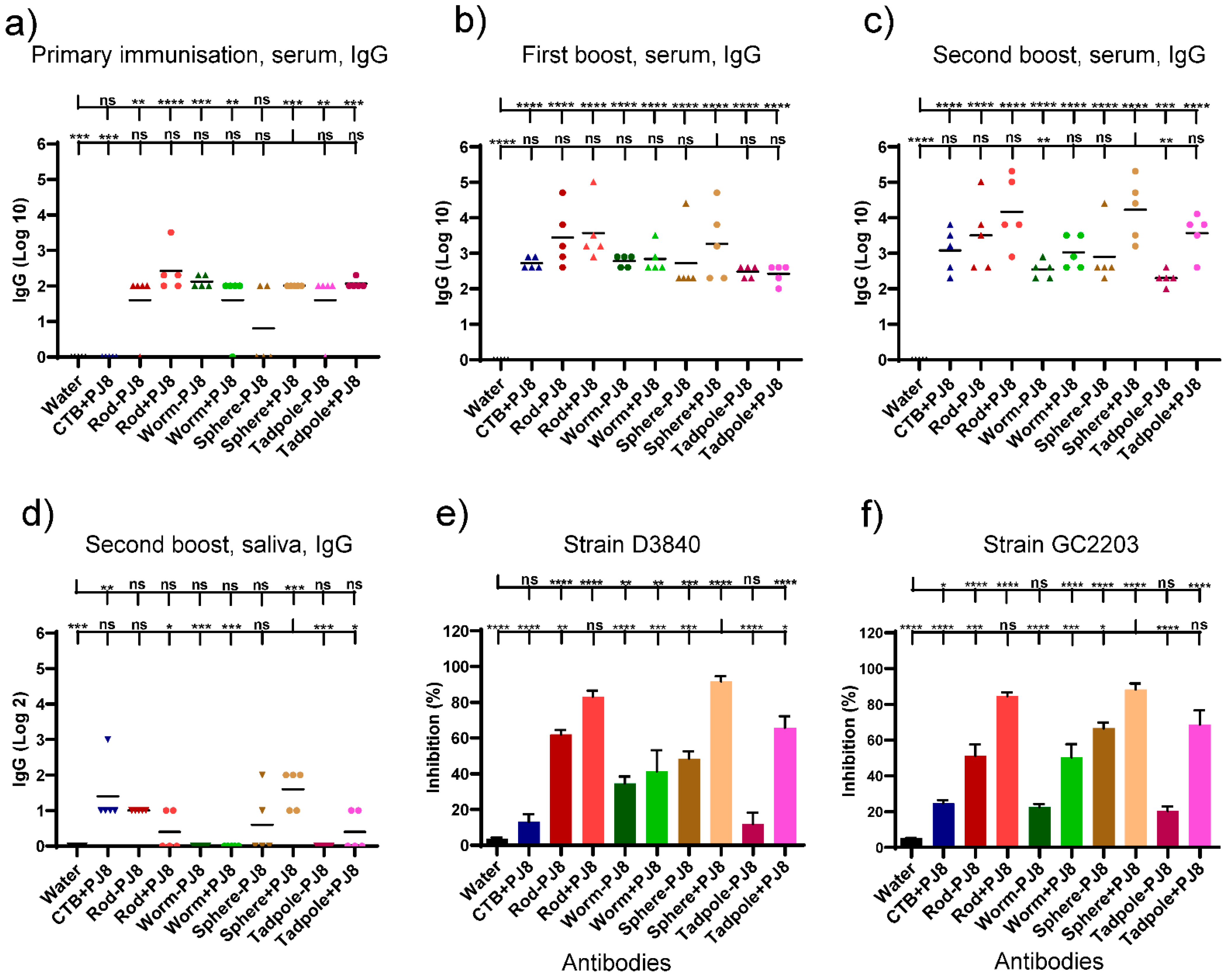
2.2. Evaluation of Antibody Responses upon Oral Vaccine Delivery
2.3. Evaluation of Antibody Responses upon Nasal Vaccine Delivery
2.4. Correlates of Immunity
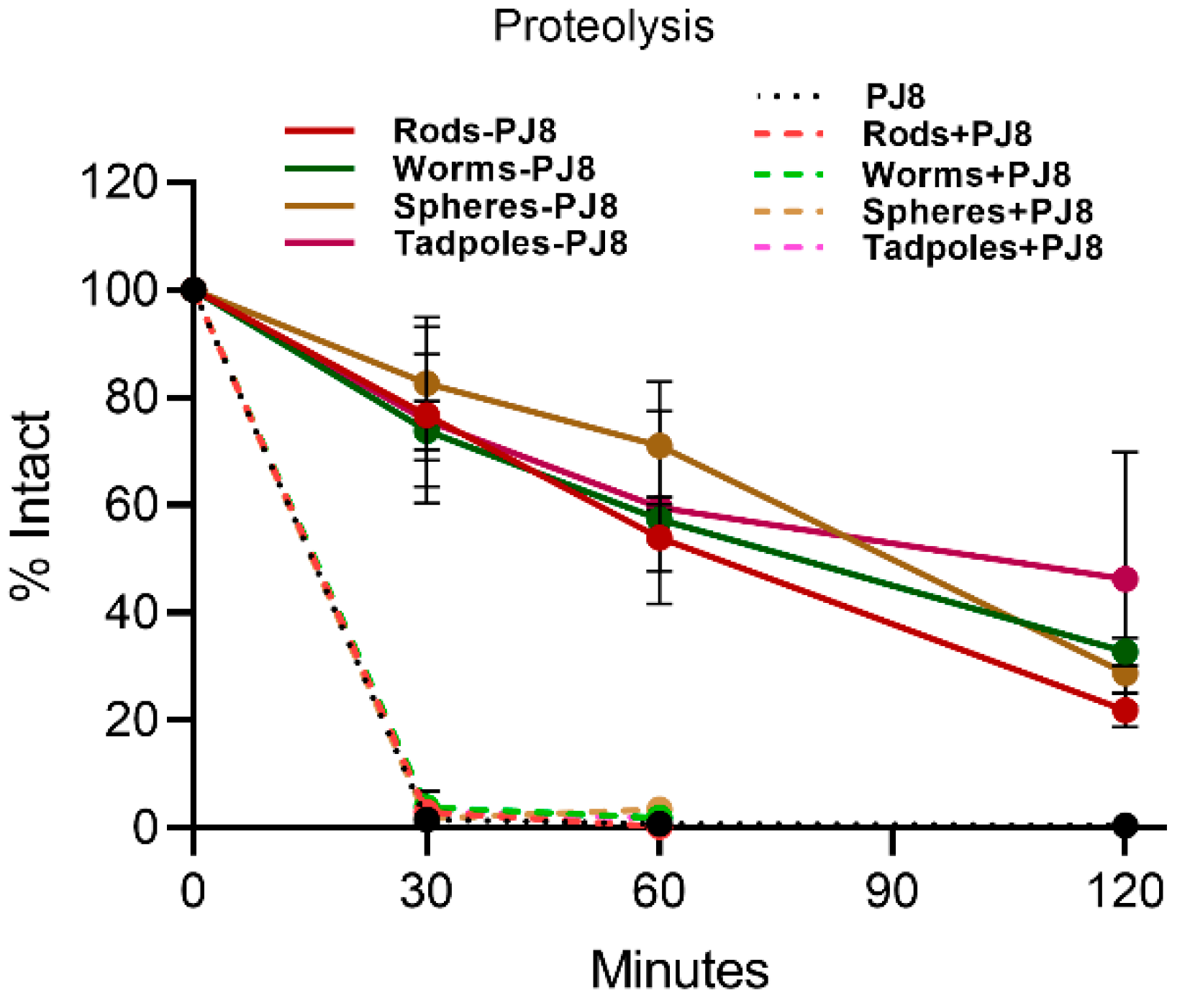
2.5. Enzymatic Stability In Vitro
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Materials and Equipment
5.2. Synthesis of Peptide Antigen
5.3. Synthesis of Rods–PJ8 and Worms–PJ8
5.4. Synthesis of Spheres–PJ8 and Tadpoles–PJ8
5.5. Preparation of Physical Mixtures of Polymeric Nanoparticles and PJ8
5.6. Characterization of Vaccine Candidates
5.7. Immunological Assessment
5.8. Oral Immunization Study
5.9. Intranasal Immunization Study
5.10. Determination of Antibody Titers
5.11. Indirect Bactericidal Assay
5.12. In Vitro Stability against Proteolysis
5.13. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- Shakya, A.K.; Chowdhury, M.Y.E.; Tao, W.; Gill, H.S. Mucosal vaccine delivery: Current state and a pediatric perspective. J. Control. Release 2016, 240, 394–413. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Non-invasive mucosal vaccine delivery: Advantages, challenges and the future. Expert Opin. Drug Deliv. 2020, 17, 435–437. [Google Scholar] [CrossRef]
- Vela Ramirez, J.E.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, S.; Rivera-Hernandez, T.; Curren, B.F.; Harbison-Price, N.; De Oliveira, D.M.P.; Jespersen, M.G.; Davies, M.R.; Walker, M.J. Pathogenesis, epidemiology and control of Group A Streptococcus infection. Nat. Rev. Microbiol. 2023, 21, 431–447. [Google Scholar] [CrossRef]
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.M.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A.; et al. Global, Regional, and National Burden of Rheumatic Heart Disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yu, D.; Yang, Y. Group A Streptococcus Is Still at Large. J. Clin. Med. 2023, 12, 2739. [Google Scholar] [CrossRef]
- Bagcchi, S. Surge of invasive Group A Streptococcus disease. Lancet Infect. Dis. 2023, 23, 284. [Google Scholar] [CrossRef]
- Barnes, M.; Youngkin, E.; Zipprich, J.; Bilski, K.; Gregory, C.J.; Dominguez, S.R.; Mumm, E.; McMahon, M.; Como-Sabetti, K.; Lynfield, R.; et al. Notes from the Field: Increase in Pediatric Invasive Group A Streptococcus Infections—Colorado and Minnesota, October-December 2022. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 265–267. [Google Scholar] [CrossRef]
- Davies, M.R.; Keller, N.; Brouwer, S.; Jespersen, M.G.; Cork, A.J.; Hayes, A.J.; Pitt, M.E.; De Oliveira, D.M.P.; Harbison-Price, N.; Bertolla, O.M.; et al. Detection of Streptococcus pyogenes M1(UK) in Australia and characterization of the mutation driving enhanced expression of superantigen SpeA. Nat. Commun. 2023, 14, 1051. [Google Scholar] [CrossRef]
- Azuar, A.; Jin, W.; Mukaida, S.; Hussein, W.; Toth, I.; Skwarczynski, M. Recent advances in the development of peptide vaccines and their delivery systems against Group A Streptococcus. Vaccines 2019, 7, 58. [Google Scholar] [CrossRef]
- Koirala, P.; Chen, S.-P.R.; Boer, J.C.; Khalil, Z.G.; Deceneux, C.; Goodchild, G.; Lu, L.; Faruck, M.O.; Shalash, A.O.; Bashiri, S.; et al. Polymeric Nanoparticles as a Self-Adjuvanting Peptide Vaccine Delivery System: The Role of Shape. Adv. Funct. Mater. 2023, 33, 2209304. [Google Scholar] [CrossRef]
- Hayman, W.A.; Brandt, E.R.; Relf, W.A.; Cooper, J.; Saul, A.; Good, M.F. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A Streptococcus. Int. Immunol. 1997, 9, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; del Guercio, M.F.; Maewal, A.; Qiao, L.; Fikes, J.; Chesnut, R.W.; Paulson, J.; Bundle, D.R.; DeFrees, S.; Sette, A. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J. Immunol. 2000, 164, 1625–1633. [Google Scholar] [CrossRef]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O’Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A.; et al. Evaluation of safety and immunogenicity of a group A Streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef]
- La Rosa, C.; Longmate, J.; Lacey, S.F.; Kaltcheva, T.; Sharan, R.; Marsano, D.; Kwon, P.; Drake, J.; Williams, B.; Denison, S.; et al. Clinical evaluation of safety and immunogenicity of PADRE-cytomegalovirus (CMV) and tetanus-CMV fusion peptide vaccines with or without PF03512676 adjuvant. J. Infect. Dis. 2012, 205, 1294–1304. [Google Scholar] [CrossRef]
- Georgousakis, M.M.; McMillan, D.J.; Batzloff, M.R.; Sriprakash, K.S. Moving forward: A mucosal vaccine against group A Streptococcus. Expert Rev. Vaccines 2009, 8, 747–760. [Google Scholar] [CrossRef]
- Shalash, A.O.; Becker, L.; Yang, J.; Giacomin, P.; Pearson, M.; Hussein, W.M.; Loukas, A.; Skwarczynski, M.; Toth, I. Oral Peptide Vaccine against Hookworm Infection: Correlation of Antibody Titers with Protective Efficacy. Vaccines 2021, 9, 1034. [Google Scholar] [CrossRef]
- Facciola, A.; Visalli, G.; Lagana, P.; La Fauci, V.; Squeri, R.; Pellicano, G.F.; Nunnari, G.; Trovato, M.; Di Pietro, A. The new era of vaccines: The “nanovaccinology”. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7163–7182. [Google Scholar] [CrossRef]
- Bartlett, S.; Eichenberger, R.M.; Nevagi, R.J.; Ghaffar, K.A.; Marasini, N.; Dai, Y.; Loukas, A.; Toth, I.; Skwarczynski, M. Lipopeptide-Based Oral Vaccine Against Hookworm Infection. J. Infect. Dis. 2020, 221, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Ran, F.; Cui, Y.; Liu, C.; Zhao, Q.; Gao, Y.; Wang, D.; Wang, S. A comparison between sphere and rod nanoparticles regarding their in vivo biological behavior and pharmacokinetics. Sci. Rep. 2017, 7, 4131. [Google Scholar] [CrossRef]
- Yu, M.; Wang, J.; Yang, Y.; Zhu, C.; Su, Q.; Guo, S.; Sun, J.; Gan, Y.; Shi, X.; Gao, H. Rotation-Facilitated Rapid Transport of Nanorods in Mucosal Tissues. Nano Lett. 2016, 16, 7176–7182. [Google Scholar] [CrossRef]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [PubMed]
- George Chandy, A.; Hultkrantz, S.; Raghavan, S.; Czerkinsky, C.; Lebens, M.; Telemo, E.; Holmgren, J. Oral tolerance induction by mucosal administration of cholera toxin B-coupled antigen involves T-cell proliferation in vivo and is not affected by depletion of CD25+ T cells. Immunology 2006, 118, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Faruck, M.O.; Zhao, L.; Hussein, W.M.; Khalil, Z.G.; Capon, R.J.; Skwarczynski, M.; Toth, I. Polyacrylate–Peptide Antigen Conjugate as a Single-Dose Oral Vaccine against Group A Streptococcus. Vaccines 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Faruck, M.O.; Koirala, P.; Yang, J.; D’Occhio, M.J.; Skwarczynski, M.; Toth, I. Polyacrylate-GnRH Peptide Conjugate as an Oral Contraceptive Vaccine Candidate. Pharmaceutics 2021, 13, 1081. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, H.; Kett, V. Current prospects and future challenges for nasal vaccine delivery. Hum. Vaccines Immunother. 2017, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.A.; Paul, S.; Dellosa, G.K.Y.; Jaraquemada, D.; Bello, M.B. Towards the future exploration of mucosal mRNA vaccines against emerging viral diseases; lessons from existing next-generation mucosal vaccine strategies. npj Vaccines 2022, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Anggraeni, R.; Ana, I.D.; Wihadmadyatami, H. Development of mucosal vaccine delivery: An overview on the mucosal vaccines and their adjuvants. Clin. Exp. Vaccine Res. 2022, 11, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Sinani, G.; Sessevmez, M.; Gök, M.K.; Özgümüş, S.; Alpar, H.O.; Cevher, E. Modified chitosan-based nanoadjuvants enhance immunogenicity of protein antigens after mucosal vaccination. Int. J. Pharm. 2019, 569, 118592. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, X.; Zhang, C.; Shao, X.; Zhang, X.; Zhang, Q.; Jiang, X. Conjugating influenza a (H1N1) antigen to n-trimethylaminoethylmethacrylate chitosan nanoparticles improves the immunogenicity of the antigen after nasal administration. J. Med. Virol. 2015, 87, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Slütter, B.; Bal, S.M.; Que, I.; Kaijzel, E.; Löwik, C.; Bouwstra, J.; Jiskoot, W. Antigen–Adjuvant Nanoconjugates for Nasal Vaccination: An Improvement over the Use of Nanoparticles? Mol. Pharm. 2010, 7, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.F.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for group A Streptococcus (GAS) vaccine. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jin, W.; Cruz, J.G.; Marasini, N.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; Skwarczynski, M.; Toth, I. Development of polyelectrolyte complexes for the delivery of peptide-based subunit vaccines against group A Streptococcus. Nanomaterials 2020, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Chandrudu, S.; Toth, I. Strategies for intranasal delivery of vaccines. Drug Deliv. Transl. Res. 2013, 3, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Bobrin, V.A.; Chen, S.-P.R.; Wang, Z.; Schoning, J.P.; Gu, Y.; Chen, W.; Chen, M.; Jia, Z.; Monteiro, M.J. Biodistribution of PNIPAM-Coated Nanostructures Synthesized by the TDMT Method. Biomacromolecules 2019, 20, 625–634. [Google Scholar] [CrossRef]
- Jia, Z.; Bobrin, V.A.; Truong, N.P.; Gillard, M.; Monteiro, M.J. Multifunctional Nanoworms and Nanorods through a One-Step Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136, 5824–5827. [Google Scholar] [CrossRef]
- Bobrin, V.A.; Monteiro, M.J. Temperature-Directed Self-Assembly of Multifunctional Polymeric Tadpoles. J. Am. Chem. Soc. 2015, 137, 15652–15655. [Google Scholar] [CrossRef]
- Perera, G.; Greindl, M.; Palmberger, T.F.; Bernkop-Schnürch, A. Insulin-loaded poly(acrylic acid)-cysteine nanoparticles: Stability studies towards digestive enzymes of the intestine. Drug Deliv. 2009, 16, 254–260. [Google Scholar] [CrossRef]
- Ansari, M.J.; Anwer, M.K.; Jamil, S.; Al-Shdefat, R.; Ali, B.E.; Ahmad, M.M.; Ansari, M.N. Enhanced oral bioavailability of insulin-loaded solid lipid nanoparticles: Pharmacokinetic bioavailability of insulin-loaded solid lipid nanoparticles in diabetic rats. Drug Deliv. 2016, 23, 1972–1979. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koirala, P.; Shalash, A.O.; Chen, S.-P.R.; Faruck, M.O.; Wang, J.; Hussein, W.M.; Khalil, Z.G.; Capon, R.J.; Monteiro, M.J.; Toth, I.; et al. Polymeric Nanoparticles as Oral and Intranasal Peptide Vaccine Delivery Systems: The Role of Shape and Conjugation. Vaccines 2024, 12, 198. https://doi.org/10.3390/vaccines12020198
Koirala P, Shalash AO, Chen S-PR, Faruck MO, Wang J, Hussein WM, Khalil ZG, Capon RJ, Monteiro MJ, Toth I, et al. Polymeric Nanoparticles as Oral and Intranasal Peptide Vaccine Delivery Systems: The Role of Shape and Conjugation. Vaccines. 2024; 12(2):198. https://doi.org/10.3390/vaccines12020198
Chicago/Turabian StyleKoirala, Prashamsa, Ahmed O. Shalash, Sung-Po R. Chen, Mohammad O. Faruck, Jingwen Wang, Waleed M. Hussein, Zeinab G. Khalil, Robert J. Capon, Michael J. Monteiro, Istvan Toth, and et al. 2024. "Polymeric Nanoparticles as Oral and Intranasal Peptide Vaccine Delivery Systems: The Role of Shape and Conjugation" Vaccines 12, no. 2: 198. https://doi.org/10.3390/vaccines12020198
APA StyleKoirala, P., Shalash, A. O., Chen, S.-P. R., Faruck, M. O., Wang, J., Hussein, W. M., Khalil, Z. G., Capon, R. J., Monteiro, M. J., Toth, I., & Skwarczynski, M. (2024). Polymeric Nanoparticles as Oral and Intranasal Peptide Vaccine Delivery Systems: The Role of Shape and Conjugation. Vaccines, 12(2), 198. https://doi.org/10.3390/vaccines12020198