

Glioblastoma Vaccines as Promising Immune-Therapeutics: Challenges and Current Status

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. The Ongoing Challenges of Glioblastoma Treatment
2.1. Blood–Brain Barrier
2.2. Immunotherapy Targeting the Tumor Microenvironment
3. Vaccine Therapy
3.1. Peptide Vaccines
3.1.1. Wilms Tumor 1
3.1.2. Survivin
3.1.3. Cytomegalovirus
3.1.4. Neoantigen
3.1.5. Multipeptide Vaccines
3.2. Dendritic Cell-Based Vaccines
3.3. Biomarkers as Potential Targets for Vaccine Therapy
3.3.1. IDH
3.3.2. EGFR
3.3.3. TERT
4. Clinical Side and Limitations
5. Potential Solutions to Overcome the Immunotherapy Challenges of Glioblastoma
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lukas, R.V.; Wainwright, D.A.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncol. Williston Park 2019, 33, 91–100. [Google Scholar]
- Mrugala, M.M.; Ruzevick, J.; Zlomanczuk, P.; Lukas, R.V. Tumor Treating Fields in Neuro-Oncological Practice. Curr. Oncol. Rep. 2017, 19, 53. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Pacheco, P.; Stupp, R. Tumor treating fields: A novel treatment modality and its use in brain tumors. Neuro-Oncology 2016, 18, 1338–1349. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncology 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Desai, K.; Hubben, A.; Ahluwalia, M. The Role of Checkpoint Inhibitors in Glioblastoma. Target. Oncol. 2019, 14, 375–394. [Google Scholar] [CrossRef]
- Minniti, G.; Muni, R.; Lanzetta, G.; Marchetti, P.; Enrici, R.M. Chemotherapy for glioblastoma: Current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009, 29, 5171–5184. [Google Scholar] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncology 2022, 24, 1318–1330. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood-Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, K.; et al. Opening of the Blood-Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Geraldo, L.H.; Garcia, C.; da Fonseca, A.C.; Dubois, L.G.; Matias, D.; de Camargo Magalhães, E.S.; Do Amaral, R.F.; da Rosa, B.G.; Grimaldi, I.; Leser, F.S.; et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 2019, 5, 46–65. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef]
- Šamec, N.; Zottel, A.; Videtič Paska, A.; Jovčevska, I. Nanomedicine and Immunotherapy: A Step Further towards Precision Medicine for Glioblastoma. Molecules 2020, 25, 490. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Swartz, A.M.; Batich, K.A.; Fecci, P.E.; Sampson, J.H. Peptide vaccines for the treatment of glioblastoma. J. Neurooncol. 2015, 123, 433–440. [Google Scholar] [CrossRef]
- Izumoto, S.; Tsuboi, A.; Oka, Y.; Suzuki, T.; Hashiba, T.; Kagawa, N.; Hashimoto, N.; Maruno, M.; Elisseeva, O.A.; Shirakata, T.; et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J. Neurosurg. 2008, 108, 963–971. [Google Scholar] [CrossRef]
- Oji, Y.; Hashimoto, N.; Tsuboi, A.; Murakami, Y.; Iwai, M.; Kagawa, N.; Chiba, Y.; Izumoto, S.; Elisseeva, O.; Ichinohasama, R.; et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int. J. Cancer 2016, 139, 1391–1401. [Google Scholar] [CrossRef]
- Takashima, S.; Oka, Y.; Fujiki, F.; Morimoto, S.; Nakajima, H.; Nakae, Y.; Nakata, J.; Nishida, S.; Hosen, N.; Tatsumi, N.; et al. Syndecan-4 as a biomarker to predict clinical outcome for glioblastoma multiforme treated with WT1 peptide vaccine. Future Sci. OA 2016, 2, FSO96. [Google Scholar] [CrossRef]
- Fu, S.; Piccioni, D.E.; Liu, H.; Lukas, R.V.; Kesari, S.; Aregawi, D.; Hong, D.S.; Yamaguchi, K.; Whicher, K.; Zhang, Y.; et al. A phase I study of the WT2725 dosing emulsion in patients with advanced malignancies. Sci. Rep. 2021, 11, 22355. [Google Scholar] [CrossRef]
- Kawanishi, Y.; Udaka, K.; Yawata, T.; Nakai, E.; Fukuda, H.; Fukui, N.; Ueba, T. IMT-03 Clinical Trial for Newly Diagnosed Malignant Glioma with Wt1-W10 Vaccination. Neuro-Oncol. Adv. 2019, 1 (Suppl. 2), ii17. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Ciesielski, M.; Abad, A.; Reardon, D.; Aiken, R.; Barbaro, M.; Sinicrope, K.; Peereboom, D.M.; Odia, Y.; Brenner, A.; et al. P07.09.B A Randomized Phase 2b Study of Survivin Vaccine Survaxm Plus Adjuvant Temozolomide for Newly-Diagnosed Glioblastoma (Survive). Neuro-Oncology 2023, 25 (Suppl. 2), ii52–ii53. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Ctim-10. Phase ii Study of Pembrolizumab Plus Survaxm for Glioblastoma at First Recurrence. Neuro-Oncology 2020, 22 (Suppl. 2), ii34–ii35. [Google Scholar] [CrossRef]
- Ciesielski, M.J.; Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Peereboom, D.M.; Figel, S.A.; Hutson, A.; Groman, A.; et al. Final data from the phase 2a single-arm trial of SurVaxM for newly diagnosed glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2037. [Google Scholar] [CrossRef]
- Rahman, M.; Dastmalchi, F.; Karachi, A.; Mitchell, D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. OncoImmunology 2019, 8, e1514921. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.G.; Bao, L.; Bruggeman, R.; Dunham, K.; Specht, C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J. Neurooncol. 2011, 103, 231–238. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Weathers, S.-P.S.; Penas-Prado, M.; Banerjee, P.P.; Bdiwi, M.; Shaim, H.; Alsuliman, A.; Shanley, M.; Long, J.; De Groot, J.F.; O’Brien, B.J.; et al. A phase I/II clinical trial of autologous CMV-specific T cells in glioblastoma (GBM) patients to reveal a lack of immune effector function. J. Clin. Oncol. 2020, 38 (Suppl. 15), 2515. [Google Scholar] [CrossRef]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Sampson, J.H.; Batich, K.A.; Mitchell, D.A.; Herndon, J.E.; Broadwater, G.; Healy, P.; Sanchez-Perez, L.; Nair, S.; Congdon, K.; Norberg, P.; et al. Reproducibility of outcomes in sequential trials using CMV-targeted dendritic cell vaccination for glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2005. [Google Scholar] [CrossRef]
- Melnick, K.; Dastmalchi, F.; Mitchell, D.; Rahman, M.; Sayour, E.J. Contemporary RNA Therapeutics for Glioblastoma. Neuromol. Med. 2022, 24, 8–12. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hargrave, A.; Mustafa, A.S.; Hanif, A.; Tunio, J.H.; Hanif, S.N.M. Recent Advances in Cancer Immunotherapy with a Focus on FDA-Approved Vaccines and Neoantigen-Based Vaccines. Vaccines 2023, 11, 1633. [Google Scholar] [CrossRef]
- Schijns, V.E.J.C.; Pretto, C.; Devillers, L.; Pierre, D.; Hofman, F.M.; Chen, T.C.; Mespouille, P.; Hantos, P.; Glorieux, P.; Bota, D.A.; et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine 2015, 33, 2690–2696. [Google Scholar] [CrossRef]
- Arakawa, Y.; Okita, Y.; Narita, Y. Efficacy finding cohort of a cancer peptide vaccine, TAS0313, in treating recurrent glioblastoma. J. Clin. Oncol. 2021, 39 (Suppl. 15), 2038. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Wick, W.; Idbaih, A.; Tabatabai, G.; Vieito, M.; Stradella, A.; Ghiringhelli, F.; Burger, M.C.; Mildenberger, I.; Herrlinger, U.; Renovanz, M.; et al. EO2401, a novel microbiome-derived therapeutic vaccine for patients with recurrent glioblastoma: ROSALIE study. J. Clin. Oncol. 2022, 40 (Suppl. 16), 2034. [Google Scholar] [CrossRef]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Liu, J.; Meng, H.; Zhang, R.; Ma, L.; Wu, L.; Yu, S.; Shi, F.; Li, Y.; et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017, 411, 182–190. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef]
- Nishikawa, M.; Takemoto, S.; Takakura, Y. Heat shock protein derivatives for delivery of antigens to antigen presenting cells. Int. J. Pharm. 2008, 354, 23–27. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Shi, Q.; Anderson, S.K.; Knopp, M.; Raizer, J.; Clarke, J.; Waziri, A.; Colman, H.; Bruce, J.; Olson, J.J.; et al. ATIM-14. Alliance A071101: A Phase Ii Randomized Trial Comparing the Efficacy of Heat Shock Protein Peptide Complex-96 (Hsppc-96) Vaccine Given with Bevacizumab Versus Bevacizumab Alone in the Treatment of Surgically Resectable Recurrent Glioblastoma. Neuro-Oncology 2017, 19 (Suppl. 6), vi29. [Google Scholar] [CrossRef]
- Kim, C.-H.; Woo, S.-J.; Park, J.-S.; Kim, H.-S.; Park, M.-Y.; Park, S.-D.; Hong, Y.-K.; Kim, T.-G. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma. Immunology 2007, 122, 615–622. [Google Scholar] [CrossRef]
- Chen, J.-R.; Yao, Y.; Xu, H.-Z.; Qin, Z.-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients With Glioblastomas. Medicine 2016, 95, e2583. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH mutation and MGMT promoter methylation in glioblastoma: Results of a prospective registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Bai, J.; Varghese, J.; Jain, R. Adult Glioma WHO Classification Update, Genomics, and Imaging: What the Radiologists Need to Know. Top. Magn. Reson. Imaging 2020, 29, 71–82. [Google Scholar] [CrossRef]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef]
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.-A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Harmancı, A.S.; Erson-Omay, E.Z.; Li, J.; Coşkun, S.; Simon, M.; Krischek, B.; Özduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Wahner, H.C.W.; Träger, M.; Bender, K.; Schweizer, L.; Onken, J.; Senger, C.; Ehret, F.; Budach, V.; Kaul, D. Predicting survival in anaplastic astrocytoma patients in a single-center cohort of 108 patients. Radiat. Oncol. 2020, 15, 282. [Google Scholar] [CrossRef]
- Christians, A.; Adel-Horowski, A.; Banan, R.; Lehmann, U.; Bartels, S.; Behling, F.; Barrantes-Freer, A.; Stadelmann, C.; Rohde, V.; Stockhammer, F.; et al. The prognostic role of IDH mutations in homogeneously treated patients with anaplastic astrocytomas and glioblastomas. Acta Neuropathol. Commun. 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017, 19, 1460–1468. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Platten, M.; Schilling, D.; Bunse, L.; Wick, A.; Bunse, T.; Riehl, D.; Green, E.; Sanghvi, K.; Karapanagiotou-Schenkel, I.; Harting, I.; et al. ATIM-33. NOA-16: A First-in-Man Multicenter Phase I Clinical Trial of the German Neurooncology Working Group Evaluating a Mutation-Specific Peptide Vaccine Targeting Idh1r132h IN Patients with Newly Diagnosed Malignant Astrocytomas. Neuro-Oncology 2018, 20 (Suppl. 6), vi8–vi9. [Google Scholar] [CrossRef]
- Senhaji, N.; Louati, S.; Chbani, L.; El Fatemi, H.; Hammas, N.; Mikou, K.; Maaroufi, M.; Benzagmout, M.; Boujraf, S.; El Bardai, S.; et al. EGFR Amplification and IDH Mutations in Glioblastoma Patients of the Northeast of Morocco. BioMed Res. Int. 2017, 2017, 8045859. [Google Scholar] [CrossRef]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef]
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia Pac. J. Clin. Oncol. 2018, 14, 40–51. [Google Scholar] [CrossRef]
- Wong, E.; Nahar, N.; Hau, E.; Varikatt, W.; Gebski, V.; Ng, T.; Jayamohan, J.; Sundaresan, P. Cut-point for Ki-67 proliferation index as a prognostic marker for glioblastoma. Asia Pac. J. Clin. Oncol. 2019, 15, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- van den Bent, M.J.; Hartmann, C.; Preusser, M.; Ströbel, T.; Dubbink, H.J.; Kros, J.M.; von Deimling, A.; Boisselier, B.; Sanson, M.; Halling, K.C.; et al. Interlaboratory comparison of IDH mutation detection. J. Neurooncol. 2013, 112, 173–178. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef]
- Jue, T.R.; McDonald, K.L. The challenges associated with molecular targeted therapies for glioblastoma. J. Neurooncol. 2016, 127, 427–434. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 2011, 13, 324–333. [Google Scholar] [CrossRef]
- Swartz, A.M.; Li, Q.-J.; Sampson, J.H. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef]
- Sampson, J.H.; Crotty, L.E.; Lee, S.; Archer, G.E.; Ashley, D.M.; Wikstrand, C.J.; Hale, L.P.; Small, C.; Dranoff, G.; Friedman, A.H.; et al. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 7503–7508. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E., II; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III–targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Dunn, G.P.; Cloughesy, T.F.; Maus, M.V.; Prins, R.M.; Reardon, D.A.; Sonabend, A.M. Emerging immunotherapies for malignant glioma: From immunogenomics to cell therapy. Neuro-Oncology 2020, 22, 1425–1438. [Google Scholar] [CrossRef]
- Nejo, T.; Matsushita, H.; Karasaki, T.; Nomura, M.; Saito, K.; Tanaka, S.; Takayanagi, S.; Hana, T.; Takahashi, S.; Kitagawa, Y.; et al. Reduced Neoantigen Expression Revealed by Longitudinal Multiomics as a Possible Immune Evasion Mechanism in Glioma. Cancer Immunol. Res. 2019, 7, 1148–1161. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499. [Google Scholar] [CrossRef]
- Nencha, U.; Rahimian, A.; Giry, M.; Sechi, A.; Mokhtari, K.; Polivka, M.; Schmitt, Y.; Di Stefano, A.-L.; Alentorn, A.; Labussière, M.; et al. TERT promoter mutations and rs2853669 polymorphism: Prognostic impact and interactions with common alterations in glioblastomas. J. Neurooncol. 2016, 126, 441–446. [Google Scholar] [CrossRef]
- Labussière, M.; Boisselier, B.; Mokhtari, K.; Di Stefano, A.-L.; Rahimian, A.; Rossetto, M.; Ciccarino, P.; Saulnier, O.; Paterra, R.; Marie, Y.; et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology 2014, 83, 1200–1206. [Google Scholar] [CrossRef]
- Olympios, N.; Gilard, V.; Marguet, F.; Clatot, F.; Di Fiore, F.; Fontanilles, M. TERT Promoter Alterations in Glioblastoma: A Systematic Review. Cancers 2021, 13, 1147. [Google Scholar] [CrossRef]
- Mosrati, M.A.; Malmström, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.-L.; Bratthäll, C.; Strandéus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663–16673. [Google Scholar] [CrossRef]
- Arita, H.; Yamasaki, K.; Matsushita, Y.; Nakamura, T.; Shimokawa, A.; Takami, H.; Tanaka, S.; Mukasa, A.; Shirahata, M.; Shimizu, S.; et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol. Commun. 2016, 4, 79. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Shu, C.; Wang, Q.; Yan, X.; Wang, J. The TERT promoter mutation status and MGMT promoter methylation status, combined with dichotomized MRI-derived and clinical features, predict adult primary glioblastoma survival. Cancer Med. 2018, 7, 3704–3712. [Google Scholar] [CrossRef]
- Amen, A.M.; Fellmann, C.; Soczek, K.M.; Ren, S.M.; Lew, R.J.; Knott, G.J.; Park, J.E.; McKinney, A.M.; Mancini, A.; Doudna, J.A.; et al. Cancer-specific loss of TERT activation sensitizes glioblastoma to DNA damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2008772118. [Google Scholar] [CrossRef]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.I.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499. [Google Scholar] [CrossRef]
- Lee, Y.; Koh, J.; Kim, S.-I.; Won, J.K.; Park, C.-K.; Choi, S.H.; Park, S.-H. The frequency and prognostic effect of TERT promoter mutation in diffuse gliomas. Acta Neuropathol. Commun. 2017, 5, 62. [Google Scholar] [CrossRef]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.B.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, X.; Jin, K.; Zhu, J.; Wang, Y.; Xiong, S.; Mao, Y.; Zhou, L. B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells. J. Neurooncol. 2008, 89, 121–129. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef]
- Vanderbeek, A.M.; Rahman, R.; Fell, G.; Ventz, S.; Chen, T.; Redd, R.; Parmigiani, G.; Cloughesy, T.F.; Wen, P.Y.; Trippa, L.; et al. The clinical trials landscape for glioblastoma: Is it adequate to develop new treatments? Neuro-Oncology 2018, 20, 1034–1043. [Google Scholar] [CrossRef]
- Lee, E.Q.; Chukwueke, U.N.; Hervey-Jumper, S.L.; de Groot, J.F.; Leone, J.P.; Armstrong, T.S.; Chang, S.M.; Arons, D.; Oliver, K.; Verble, K.; et al. Barriers to accrual and enrollment in brain tumor trials. Neuro-Oncology 2019, 21, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.; Segall, L.; Hillyer, G.; Iwamoto, F.; Kreisl, T.; Otap, D.; Lassman, A.B. Epid-11. Identifying Barriers to Clinical Research: A Pilot Study to Improve Access and Enrollment to Neuro-Oncology Trials at Columbia University Center Medical Center (Cumc). Neuro-Oncology 2017, 19 (Suppl. 6), vi71. [Google Scholar] [CrossRef]
- Lee, E.Q.; Weller, M.; Sul, J.; Bagley, S.J.; Sahebjam, S.; van den Bent, M.; Ahluwalia, M.; Campian, J.L.; Galanis, E.; Gilbert, M.R.; et al. Optimizing eligibility criteria and clinical trial conduct to enhance clinical trial participation for primary brain tumor patients. Neuro-Oncology 2020, 22, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Reardon, D.A.; Batchelor, T.T.; Chabner, B.A.; Flaherty, K.; de Groot, J.F.; Gilbert, M.R.; Galanis, E.; et al. It is time to include patients with brain tumors in phase I trials in oncology. J. Clin. Oncol. 2011, 29, 3211–3213. [Google Scholar] [CrossRef]
- Gounder, M.M.; Nayak, L.; Sahebjam, S.; Muzikansky, A.; Sanchez, A.J.; Desideri, S.; Ye, X.; Ivy, S.P.; Nabors, L.B.; Prados, M.; et al. Evaluation of the Safety and Benefit of Phase I Oncology Trials for Patients with Primary CNS Tumors. J. Clin. Oncol. 2015, 33, 3186–3192. [Google Scholar] [CrossRef] [PubMed]
- Ventz, S.; Lai, A.; Cloughesy, T.F.; Wen, P.Y.; Trippa, L.; Alexander, B.M. Design and Evaluation of an External Control Arm Using Prior Clinical Trials and Real-World Data. Clin. Cancer Res. 2019, 25, 4993–5001. [Google Scholar] [CrossRef]
- Salvato, I.; Marchini, A. Immunotherapeutic Strategies for the Treatment of Glioblastoma: Current Challenges and Future Perspectives. Cancers 2024, 16, 1276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, C.; Ge, H.; Lin, Y.; Kang, D. Glioblastoma vaccine tumor therapy research progress. Chin. Neurosurg. J. 2022, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S. NABTT CNS Consortium Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Chuntova, P.; Downey, K.M.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front. Immunol. 2019, 9, 03062. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.C. Blood–brain barrier breakdown in septic encephalopathy and brain tumours*. J. Anat. 2002, 200, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Donnadieu, E.; Dupré, L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J. Leukoc. Biol. 2020, 108, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.; Godicelj, A.; Wucherpfennig, K.W. A Conceptual Framework for Inducing T Cell-Mediated Immunity Against Glioblastoma. Semin. Immunopathol. 2022, 44, 697–707. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qu, Y.; Cheng, L.; Yoon, C.W.; He, P.; Monther, A.; Guo, T.; Chittle, S.; Wang, Y. Engineering chimeric antigen receptor T cells for solid tumour therapy. Clin. Transl. Med. 2022, 12, e1141. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Qin, Y.; Xu, G. Enhancing CAR T-cell therapies against solid tumors: Mechanisms and reversion of resistance. Front. Immunol. 2022, 13, 1053120. [Google Scholar] [CrossRef]
- Sridhar, P.; Petrocca, F. Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers 2017, 9, 92. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 867154. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Ronsley, R.; Choe, M.; Henson, C.; Breedt, M.; Barrios-Anderson, A.; Wein, A.; Brown, C.; Beebe, A.; Kong, A.; et al. Locoregional CAR T cells for children with CNS tumors: Clinical procedure and catheter safety. Neoplasia 2023, 36, 100870. [Google Scholar] [CrossRef]
- Majd, N.; Dasgupta, P.; de Groot, J. Immunotherapy for Neuro-Oncology. In Immunotherapy; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef]
- Sahebjam, S.; Sharabi, A.; Lim, M.; Kesarwani, P.; Chinnaiyan, P. Immunotherapy and radiation in glioblastoma. J. Neurooncol. 2017, 134, 531–539. [Google Scholar] [CrossRef]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biol. Targets Ther. 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Mormino, A.; Garofalo, S. Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment. Cancers 2022, 14, 2632. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zheng, Y.; Hong, W.; Chen, X.; Li, H.; Huang, B.; Huang, Z.; Tang, H.; Geng, W. Recent Advances in Immune Cell Therapy for Glioblastoma. Front. Immunol. 2020, 11, 544563. [Google Scholar] [CrossRef]
- Oelkrug, C.; Ramage, J.M. Enhancement of T cell recruitment and infiltration into tumours. Clin. Exp. Immunol. 2014, 178, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Hu, M.; Iyer, V.; Yu, J. Blocking the PD-1/PD-L1 pathway in glioma: A potential new treatment strategy. J. Hematol. Oncol. 2017, 10, 81. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, Q.; Zhang, J.; Liu, F. Targeting Tumor-Associated Antigen: A Promising CAR-T Therapeutic Strategy for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 661606. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, J.; Ruella, M.; Houot, R. Born to survive: How cancer cells resist CAR T cell therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef]
- Salinas, R.D.; Durgin, J.S.; O’Rourke, D.M. Potential of Glioblastoma-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy. CNS Drugs 2020, 34, 127–145. [Google Scholar] [CrossRef]
- Simula, L.; Ollivier, E.; Icard, P.; Donnadieu, E. Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration? Cells 2022, 11, 1854. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.M.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther.-Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef]
- Shen, S.; Chen, L.; Liu, J.; Yang, L.; Zhang, M.; Wang, L.; Zhang, R.; Uemura, Y.; Wu, Q.; Yu, X.; et al. Current state and future of co-inhibitory immune checkpoints for the treatment of glioblastoma. Cancer Biol. Med. 2020, 17, 555–568. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Prinzing, B.L.; Gottschalk, S.M.; Krenciute, G. CAR T-cell therapy for glioblastoma: Ready for the next round of clinical testing? Expert Rev. Anticancer Ther. 2018, 18, 451–461. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T With TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 01117. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Stachelek, G.C.; Grimm, J.; Moore, J.; Huang, E.; Spoleti, N.; Redmond, K.J.; Lim, M.; Bettegowda, C.; Kleinberg, L. Tumor-Treating Field Arrays Do Not Reduce Target Volume Coverage for Glioblastoma Radiation Therapy. Adv. Radiat. Oncol. 2020, 5, 62–69. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Germano, I.M.; Fable, J.; Humayun Gultekin, S.; Silvers, A. Adenovirus/Herpes Simplex-Thymidine Kinase/Ganciclovir Complex: Preliminary Results of a Phase I trial in Patients with Recurrent Malignant Gliomas. J. Neurooncol. 2003, 65, 279–289. [Google Scholar] [CrossRef]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 Clinical Trial of Intratumoral Reovirus Infusion for the Treatment of Recurrent Malignant Gliomas in Adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Ramos, M.; García-Foncillas, J. CAR-T cell and Personalized Medicine. In Translational Research and Onco-Omics Applications in the Era of Cancer Personal Genomics; Ruiz-Garcia, E., Astudillo-de la Vega, H., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; pp. 131–145. [Google Scholar] [CrossRef]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Jiang, H.; Gao, H.; Kong, J.; Song, B.; Wang, P.; Shi, B.; Wang, H.; Li, Z. Selective Targeting of Glioblastoma with EGFRvIII/EGFR Bitargeted Chimeric Antigen Receptor T Cell. Cancer Immunol. Res. 2018, 6, 1314–1326. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Elahi, R.; Khosh, E.; Esmaeilzadeh, A. Programmable and multi-targeted CARs: A new breakthrough in cancer CAR-T cell therapy. Clin. Transl. Oncol. 2021, 23, 1003–1019. [Google Scholar] [CrossRef]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef]
- Gisina, A.; Kholodenko, I.; Kim, Y.; Abakumov, M.; Lupatov, A.; Yarygin, K. Glioma Stem Cells: Novel Data Obtained by Single-Cell Sequencing. Int. J. Mol. Sci. 2022, 23, 14224. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef]
- Qin, V.M.; Haynes, N.M.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. CAR-T Plus Radiotherapy: A Promising Combination for Immunosuppressive Tumors. Front. Immunol. 2022, 12, 813832. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Shi, J.; Liu, J.; Li, Q.; Chen, L. Combination therapy: A feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol. Lett. 2018, 16, 2063–2070. [Google Scholar] [CrossRef]
- Joudeh, N.; Linke, D. Nanoparticle classification, physicochemical properties, characterization, and applications: A comprehensive review for biologists. J. Nanobiotechnol. 2022, 20, 262. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, Y.; Xu, H.; Li, L.; Qian, F.; Wang, L.; Quan, A.; Ma, H.; Liu, H.; Yu, R. Cascade-Responsive 2-DG Nanocapsules Encapsulate aV-siCPT1C Conjugates to Inhibit Glioblastoma through Multiple Inhibition of Energy Metabolism. ACS Appl. Mater. Interfaces 2023, 15, 10356–10370. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Q.Y.; Haqqani, A.S.; Leclerc, S.; Liu, Z.; Fauteux, F.; Baumann, E.; Delaney, C.E.; Ly, D.; Star, A.T.; et al. Differential expression of receptors mediating receptor-mediated transcytosis (RMT) in brain microvessels, brain parenchyma and peripheral tissues of the mouse and the human. Fluids Barriers CNS 2020, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Rip, J.; van Kregten, J.; van den Heuvel, A.; van der Pol, S.; van der Boom, B.; Reijerkerk, A.; Chen, L.; de Boer, M.; Gaillard, P.; et al. Glutathione conjugation dose-dependently increases brain-specific liposomal drug delivery in vitro and in vivo. Drug Discov. Today Technol. 2016, 20, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Vanbilloen, W.J.F.; Rechberger, J.S.; Anderson, J.B.; Nonnenbroich, L.F.; Zhang, L.; Daniels, D.J. Nanoparticle Strategies to Improve the Delivery of Anticancer Drugs across the Blood-Brain Barrier to Treat Brain Tumors. Pharmaceutics 2023, 15, 1804. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood–brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Eckert, M.A.; Riazifar, H.; Kang, D.-K.; Agalliu, D.; Zhao, W. From Blood to the Brain: Can Systemically Transplanted Mesenchymal Stem Cells Cross the Blood-Brain Barrier? Stem Cells Int. 2013, 2013, e435093. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Mao, J.; Duan, X.; Lu, L.; Zhang, F.; Lin, B.; Chen, M.; Zheng, C.; Zhang, X.; Shen, J. In vivo tracking of the tropism of mesenchymal stem cells to malignant gliomas using reporter gene-based MR imaging. Int. J. Cancer 2018, 142, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13, 1536. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Chang, Y.-H.; Rajesh, R. Targeted delivery of etoposide, carmustine and doxorubicin to human glioblastoma cells using methoxy poly(ethylene glycol)-poly(ε-caprolactone) nanoparticles conjugated with wheat germ agglutinin and folic acid. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 96, 114–128. [Google Scholar] [CrossRef]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients with Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Guan, Y.; Yoshimoto, K.; Hata, N.; Amano, T.; Nakamizo, A.; Sasaki, T. MicroRNAs in Human Malignant Gliomas. J. Oncol. 2012, 2012, e732874. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Guan, Y.; Yoshimoto, K.; Hata, N.; Amano, T.; Nakamizo, A.; Sasaki, T. Clinical implications of microRNAs in human glioblastoma. Front. Oncol. 2013, 3, 19. [Google Scholar] [CrossRef]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
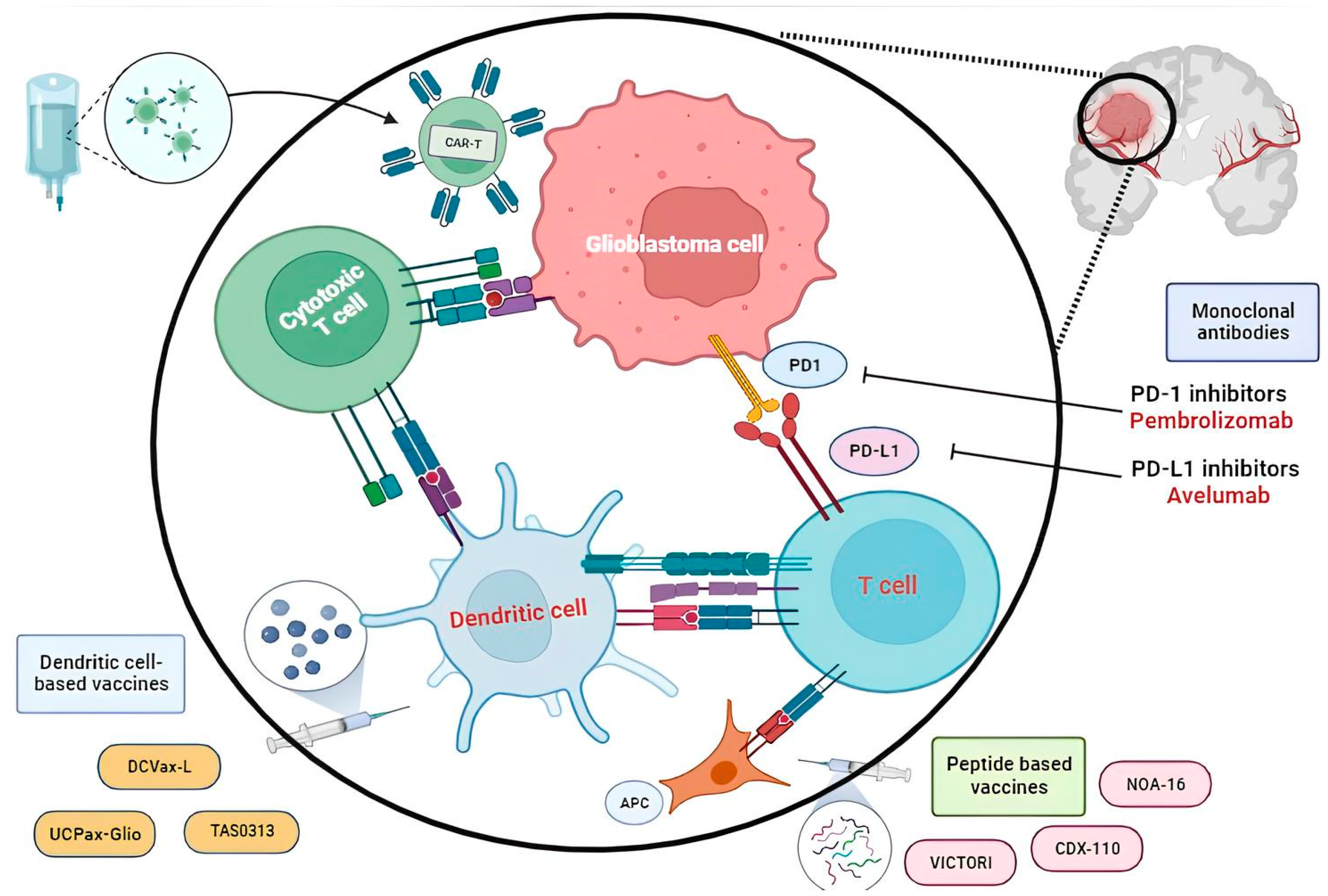

{kind=link}
| Vaccine | Trial Title | Phase | N | Experience Details | Endpoints | Clinical Trial |
|---|---|---|---|---|---|---|
| Wilms tumor 1 (WT1) | WT1 peptide vaccination for patients with recurrent glioblastoma multiforme (rGBM) | 2 | 21 | WT1 peptide | Median progression-free survival (mPFS) = 20 weeks at 6 months (mo) (26-week), PFS rate = 33% | NA (Not available) |
| WT2725 in patients with advanced malignancies | 1 | 64 | WT1 peptide (WT2725) | Response rate in GBM = 20% OS at 12 mo in GBM = 33% | NCT01621542 Completed | |
| IMT03 study for Newly Diagnosed (Nd) Malignant Glioma with WT1-W10 Vaccination | 1/2 | 27 | WT peptide (W10) | OS = 21.9 mo PFS = 12.7 mo | NA | |
| INO-5401 and INO-9012 vaccines delivered by Electroporation (EP) and combined with Cemiplimab (REGN2810) in Nd GBM | 1/2 | 52 | O6 methylguanine DNA methyltransferase (MGMT) INO-5401 + INO-9012 + Cemiplimab + RT + temozolomide (TMZ) | mOS in MGMT-UN (Unmethylated: 17.9 months vs. 32.5 months in MGMT-M (Methylated) | NCT03491683 Active, not recruiting | |
| Survivin | Evaluation of the Safety and Efficacy of SVN53-67/M57-KLH (keyhole limpet hemocyanin) (SurVaxM) in Survivin Positive Newly Diagnosed Glioblastoma | 2 | 66 | SurVaxM + TMZ | PFS at 6 mo in patients treated with at least four doses of peptide vaccine | NCT02455557 Active, not recruiting |
| Pembrolizumab Plus SurVaxM for Glioblastoma at First Recurrence | 2 | 40 | SurVaxM + Pembrolizumab | PFS at 6 mo | NCT04013672 Active, not recruiting | |
| CMV (cytomegalovirus) | Vaccine Therapy for the Treatment of Nd GBM (ATTAC-I) | 1 | 42 | pp65-DC vaccine + tetanus toxoid (Td) preconditioning | Safety and feasability OS = 20.6–47.3 mo vs. 13.8–41.3 mo (p = 0.013) | NCT00639639 Completed |
| Vaccine Therapy for the Treatment of Newly Diagnosed Glioblastoma (ATTAC- II) | 2 | 175 | CMV pp65-mRNA loaded DCs + GMCSF + Td + TMZ | mPFS = 31.4 mo and mOS = 34 mo | NCT02465268 Completed | |
| Assessment of surmounting limited migration and improving CMV specific DC Vaccines with Adjuvant Tetanus in Nd GBM (ELEVATE) | 2 | 64 | pp65 DC vaccine + TMZ vs. pp65 DC vaccine + TMZ + preconditioning | 3-year OS = 34% (95% confidence interval (CI) 19–63%) vs. 6% (95% CI 1–42%) | NCT02366728 Completed | |
| Neoantigens | Personalized NeoAntigen Cancer Vaccine With Radiotherapy (RT) Plus Pembrolizumab/MK-3475 Among Newly Diagnosed Glioblastoma Patients | 1/1b | 56 | NeoVax + RT vs. Neovax + RT + Pembrolizumab | Safety and tolerability | NCT02287428 Recruiting |
| Multipeptide Vaccines | Evaluation of the efficacy of TAS0313 in rGBM | 1/2 | 17 | TAS0313 | Safety, tolerability and efficacy. mPFS = 2.2 (95% CI, 1.0–2.3) months overall response rate (ORR) = 11.1% (95% CI = 0.3–48.2%) | JapicCTI183824 |
| Evaluation of the safety and efficacy of ICT-107 in Nd GBM | 2b | 124 | Resection + Chemoradiation + ICT107 vs. placebo | OS = 17 vs. 15 mo hazard ratio (HR) = 0.87; p = 0.58) PFS = 11.2 vs. 9.0 mo (HR = 0.57; p = 0.011) | NCT01280552 Completed | |
| First-in-Human, a Multipeptide Therapeutic Vaccine in Patients with Progressive Glioblastoma (ROSALIE) | 1b/2a | 52 | EO2041 + nivolumab vs. EO2041 + nivolumab + bevacizumab | Safety and tolerability | NCT04116658 Active, not recruiting | |
| Dendritic cells | Clinical Trial Evaluating DCVax®-L, Autologous Dendritic Cells Pulsed With Tumor Lysate Antigen to treat newly diagnosed GBM | 3 | 348 | TMZ + DCVax-L vs. TMZ + placebo | mOS = 23.1 mo (95% CI 21.2–25.4) | NCT00045968 Active, not recruiting |
| Study of a Dendritic Cell Vaccine for Patients with Either Newly Diagnosed or Recurrent GBM | 1 | 39 | DC vaccine + GBM stem-like cell lysate | Safety and tolerability mPFS = 8.75 mo in newly diagnosed GBM, 3.23 mo in recurrent GBM mOS = 20.36 mo in newly diagnosed GBM, 11.97 mo in recurrent GBM | NCT02010606 Completed | |
| Study of DC-Based Therapy Targeting Tumor Stem Cells in patients with GBM and receiving therapy | 1/2 | 20 | DC vaccine pulsed with mRNA from glioma stem cells (GSCs) | Safe study, immune response towards the stem-cell like part mOS for treated group: 759 days vs. 585 days for control group progression-free survival 694 days vs. 236 days for unvaccinated patients | NCT00846456 Completed | |
| HSP | Heat Shock Protein Peptide Complex-96 (HSPPC-96) Vaccine for cases with Recurrent or progressive High Grade Glioma | 1/2 | 96 | HSPPC-96 vaccine | OS at 6 mo = 90.2% (95% CI 75.9–96.8%) mOS = 42.6 weeks (95% CI = 34.7–50.5) | NCT00293423 Completed |
| Randomized Trial assessing the Efficacy of HSPPC-96 Vaccine in the Treatment of rGBM | 2 | 90 | HSPPC-96 vaccine+ bevacicumab compared to Bevacizumab alone | OS = 7.5 vs. 10.7 mo (HR = 2.06) | NCT01814813 Completed | |
| IDH | Targeting IDH1R132H in Glioma (WHO Grade III-IV) with IDH1R132H by a Peptide Vaccine Multicenter Trial (NOA-16) | 1 | 39 | IDH1 peptide vaccine | Safety, tolerability, and immunogenicity. Vaccine-induced immune response in 93% | NCT02454634 Completed |
| Epidermal Growth Factor Receptor Variant III (EGFRvIII) | Rindopepimut/Granulocyte Macrophage Colony Stimulating Factor (GM-CSF) In cases With Nd | 3 | 745 | TMZ vs. Rindopepimut + TMZ | OS = 20.1 (95% CI 18.5–22.1) vs. 20.0 m (95% CI 18.1–21.9) | NCT01480479 Completed |
| GBM (ACTIVATe IV) | 2 | 73 | Rindopepimut + bevacizumab vs. Bevacizumab | PFS at 6 m = 28% vs. 16% (HR = 0.72, 95% CI 0.42–1.21) | NCT01498328 Completed | |
| Rindopepimut /GM-CSF in cases with EGFRvIII-Positive rGBM (ReACT) | ||||||
| TERT | Anticancer Therapeutic by Telomerase-derived Universal Cancer Peptides vaccine in GBM (UCPVax-Glio) | 1/2 | 56 | UCPVax +TMZ vs. UCPVax | Anti-TERT T-cell response | NCT04280848 Active, not recruiting |
| INO-5401 and INO-9012 vaccines delivered by EP and combined with Cemiplimab (REGN2810) in Nd GBM | 1/2 | 52 | MGMT INO-5401 + INO-9012 + Cemiplimab + RT + TMZ | mOS in MGMT-UN: 17.9 months vs. 32.5 months in MGMT-M | NCT03491683 Active, not recruiting |
| Challenges | Potential Solutions |
|---|---|
| BBB and low infiltration of CAR-T cells in solid tumors [8,11,133,134,135,136,137] | Delivery of anticancer drugs across the BBB using nanoparticles (NCT00734682). |
| Chemokine receptor expression in CAR-T cells [134,138,139,140,141]. | |
| Loco-regional delivery of CAR-T cells [134,138,142,143,144,145]. | |
| Immunosuppressive TME [8,129,146,147,148,149,150,151,152,153,154,155,156,157,158] | CAR-T cells can release cytokines or antibodies inducing antibody-dependent cell-mediated cytotoxicity [149,159,160,161]. |
| Secretion of checkpoint-blocking antibodies by CAR-T cells or combination of CAR-T cell therapy with checkpoint inhibitors [156,162,163,164,165,166,167]. Elimination of co-inhibiting molecules [138,156,159,168,169,170,171,172]. Specific agonists targeting tumor-associated macrophages (TAM) [173,174,175,176]. Personalized vaccination therapy targeting neoantigens [175]. | |
| Intratumoral heterogeneity and antigen loss [105,133,159,177,178,179,180] | Multiple targeted and programmable CAR-T cells [181,182,183,184]. |
| Targeting cancer stem cells to avoid recurrence [8,149,185,186]. | |
| Polytherapeutic approaches with radiotherapy or chemotherapy [148,187,188,189,190]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Squalli Houssaini, A.; Lamrabet, S.; Nshizirungu, J.P.; Senhaji, N.; Sekal, M.; Karkouri, M.; Bennis, S. Glioblastoma Vaccines as Promising Immune-Therapeutics: Challenges and Current Status. Vaccines 2024, 12, 655. https://doi.org/10.3390/vaccines12060655
Squalli Houssaini A, Lamrabet S, Nshizirungu JP, Senhaji N, Sekal M, Karkouri M, Bennis S. Glioblastoma Vaccines as Promising Immune-Therapeutics: Challenges and Current Status. Vaccines. 2024; 12(6):655. https://doi.org/10.3390/vaccines12060655
Chicago/Turabian StyleSqualli Houssaini, Asmae, Salma Lamrabet, Jean Paul Nshizirungu, Nadia Senhaji, Mohammed Sekal, Mehdi Karkouri, and Sanae Bennis. 2024. "Glioblastoma Vaccines as Promising Immune-Therapeutics: Challenges and Current Status" Vaccines 12, no. 6: 655. https://doi.org/10.3390/vaccines12060655
APA StyleSqualli Houssaini, A., Lamrabet, S., Nshizirungu, J. P., Senhaji, N., Sekal, M., Karkouri, M., & Bennis, S. (2024). Glioblastoma Vaccines as Promising Immune-Therapeutics: Challenges and Current Status. Vaccines, 12(6), 655. https://doi.org/10.3390/vaccines12060655