
Intranasal Vaccination with Recombinant TLR2-Active Outer Membrane Vesicles Containing Sequential M2e Epitopes Protects against Lethal Influenza a Challenge

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. OMV Production and Characterization
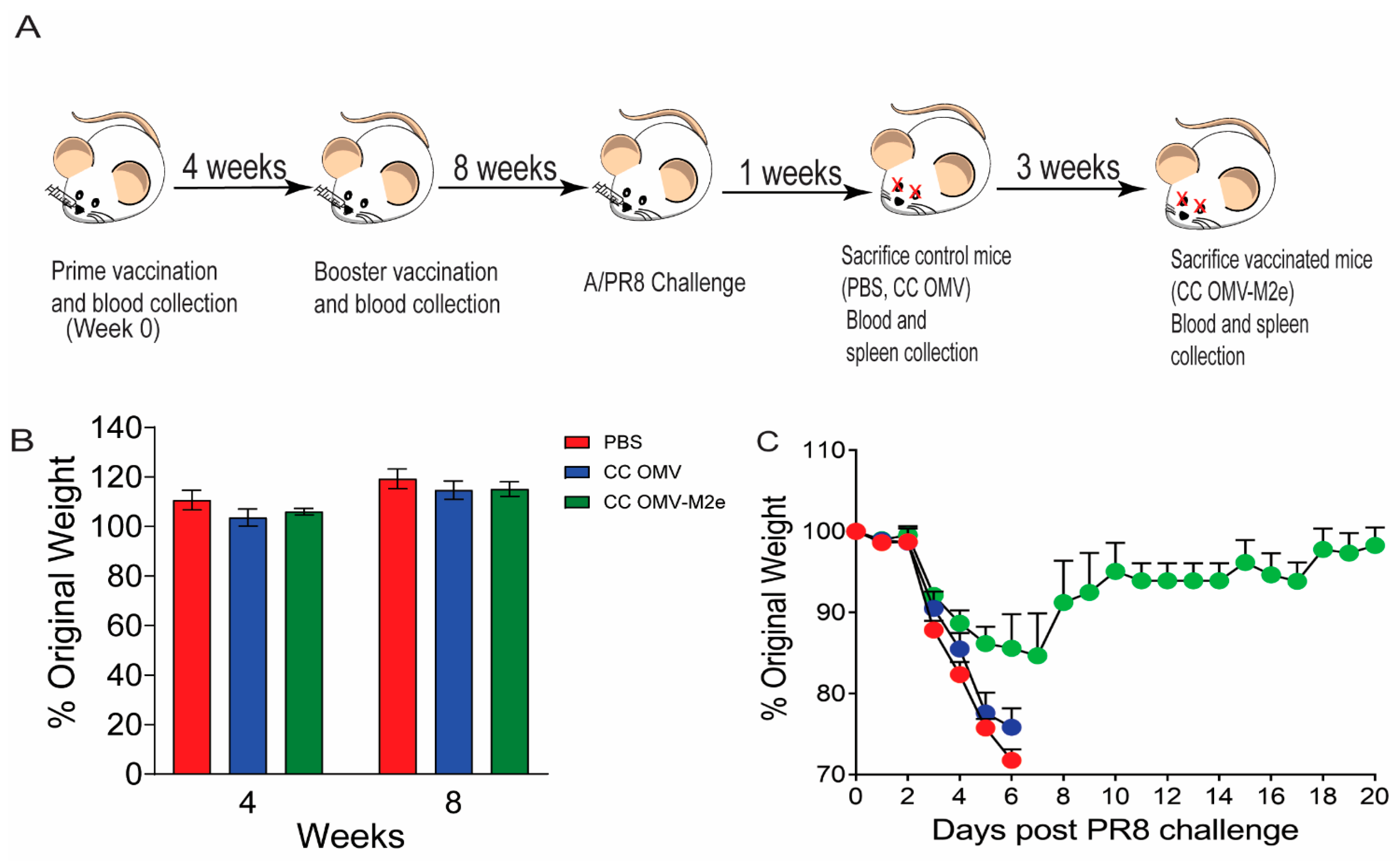
2.2. Immunization and Infection
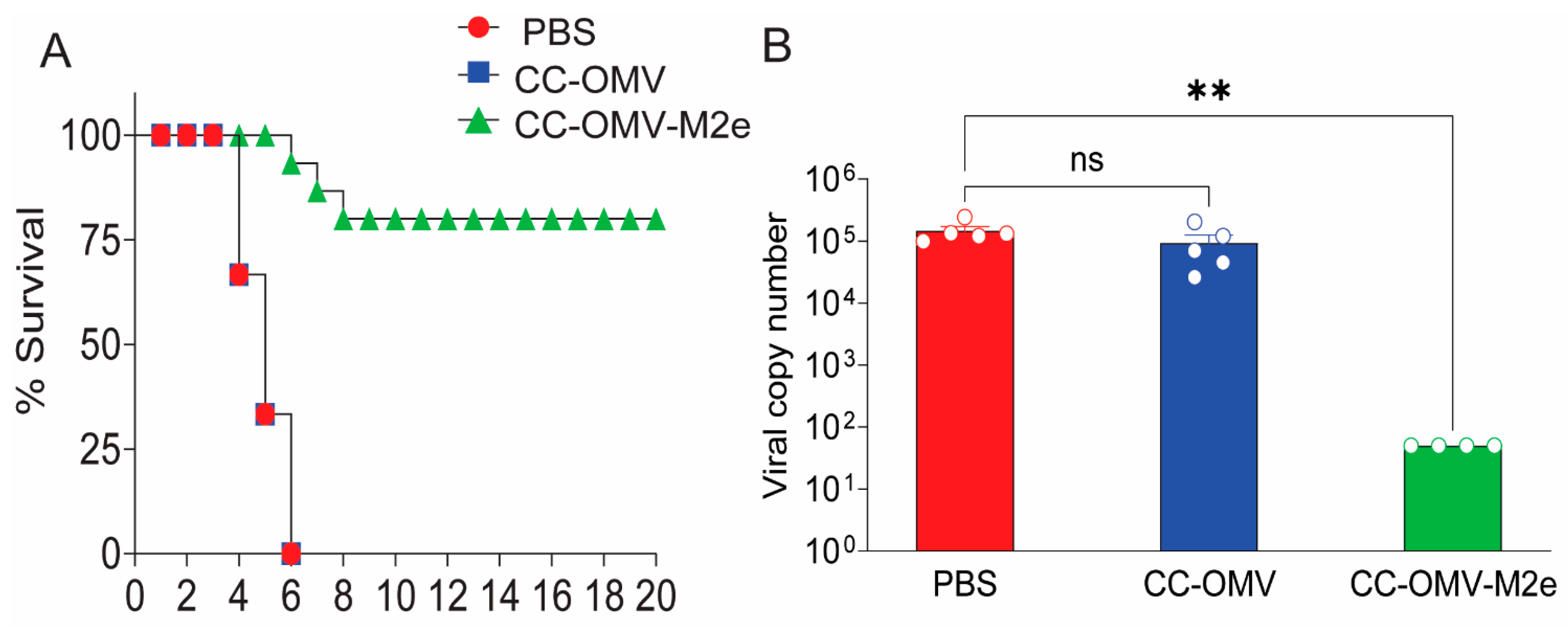
2.3. Quantification of Lung Viral Titers
2.4. Collection of Mouse Sera and Detection Serum Antibodies
2.5. Splenocyte Isolation and Culture
2.6. Flow Cytometric Analysis of Effector T Cell Cytokine
2.7. Total Cytokine Secretion Analysis Using Luminex
2.8. Statistical Analysis
3. Results
3.1. Intranasal Vaccination with CC-OMV-M2e Protects against Lethal Influenza A/PR8 Challenge in BALB/c Mice
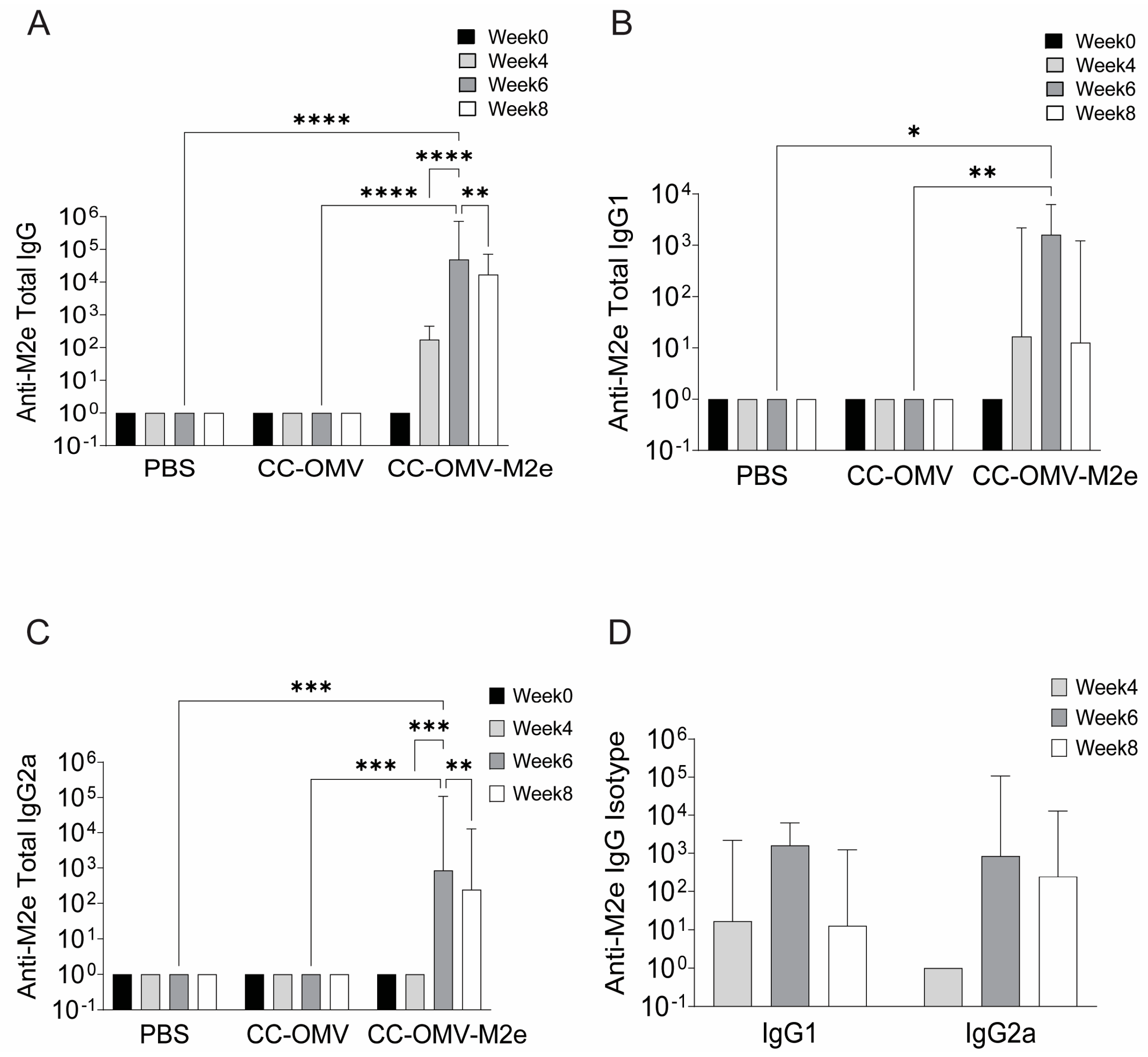
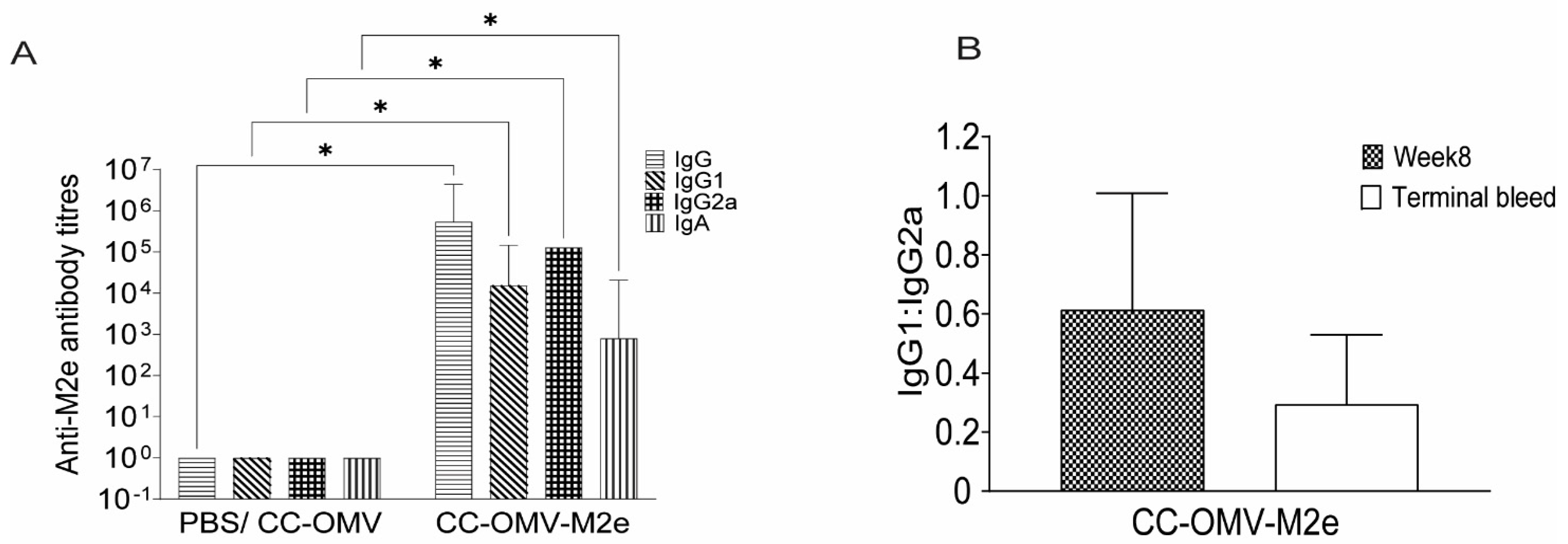
3.2. Intranasal Vaccination with CC-OMV-M2e Elicits anti-M2e IgG and IgA Titers Post-Infection and Protects Balb/C Mice from Lethal Influenza A/PR8 Challenge
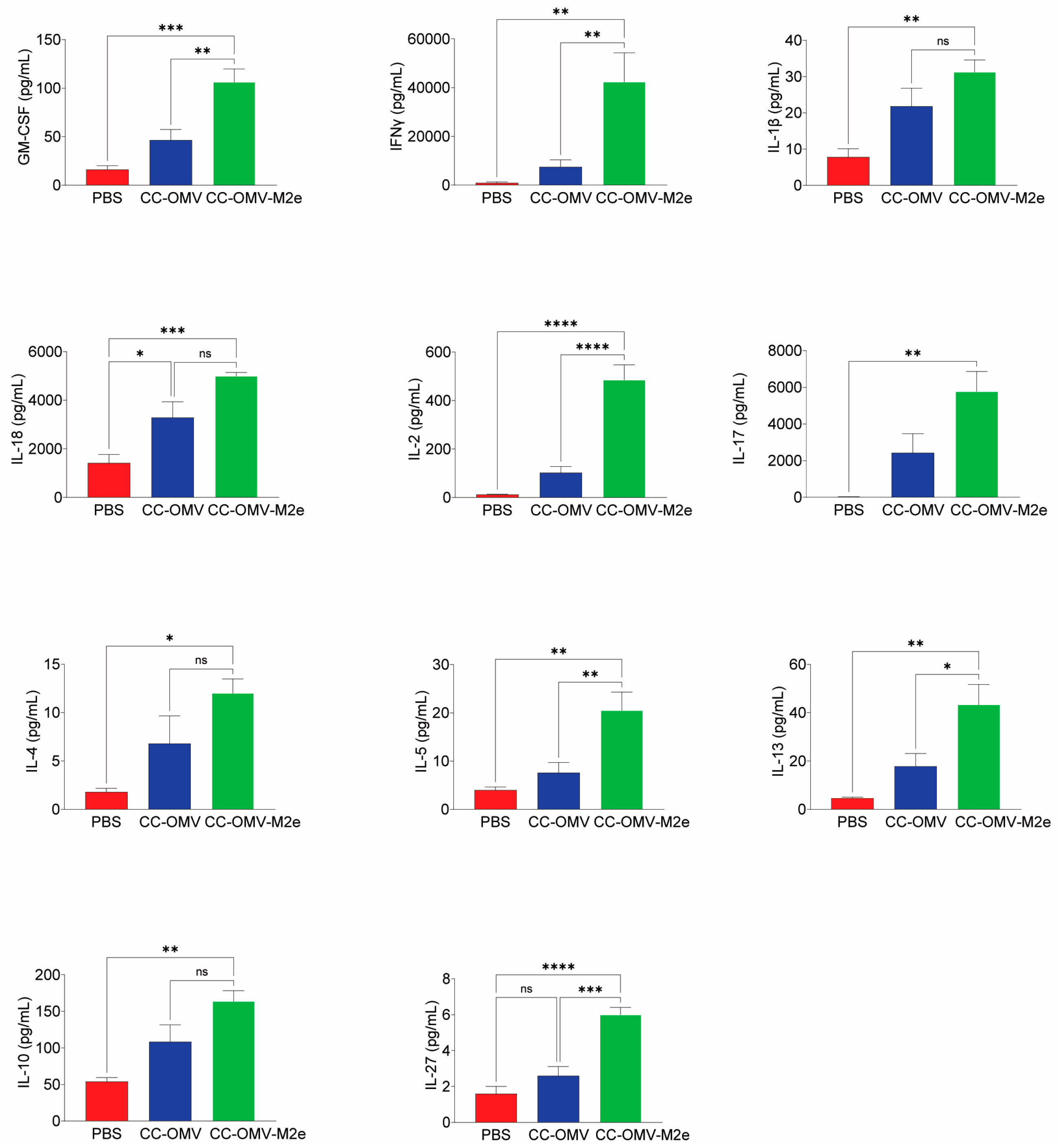
3.3. Intranasal Vaccination with CC-OMV-M2e Elicits a Th1 Bias Post-Infection and Protects Balb/c Mice against Lethal Influenza A/PR8 Challenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C. Global Mortality Associated with Seasonal Influenza Epidemics: New Burden Estimates and Predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 020421. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (U.S.). CDC Resources for PandemicFlu. Available online: https://www.cdc.gov/flu/pandemic-resources/basics/about.html (accessed on 23 June 2023).
- Treanor, J.J. Prospects for Broadly Protective Influenza Vaccines. Vaccine 2015, 33, D39–D45. [Google Scholar] [CrossRef] [PubMed]
- Schotsaert, M.; García-Sastre, A. Influenza Vaccines: A Moving Interdisciplinary Field. Viruses 2014, 6, 3809–3826. [Google Scholar] [CrossRef] [PubMed]
- Kirchenbaum, G.A.; Ross, T.M. Eliciting Broadly Protective Antibody Responses against Influenza. Curr. Opin. Immunol. 2014, 28, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Richardson, C.D.; Zebedee, S.L. Influenza Virus M2 Protein Is an Integral Membrane Protein Expressed on the Infected-Cell Surface. Virus Res. 1985, 3, 4. [Google Scholar] [CrossRef]
- Leser, G.P.; Lamb, R.A. Influenza Virus Assembly and Budding in Raft-Derived Microdomains: A Quantitative Analysis of the Surface Distribution of HA, NA and M2 Proteins. Virology 2005, 342, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martínez-Alonso, M.; Fodor, E. Conserved and Host-Specific Features of Influenza Virion Architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed]
- Khardori, N.M. Universal Vaccine Based on Ectodomain of Matrix Protein 2 of Influenza A: Fc Receptors and Alveolar Macrophages Mediate Protection. Yearb. Med. 2011, 2011, 108–109. [Google Scholar] [CrossRef]
- Jegerlehner, A.; Schmitz, N.; Storni, T.; Bachmann, M.F. Influenza A Vaccine Based on the Extracellular Domain of M2: Weak Protection Mediated via Antibody-Dependent NK Cell Activity. J. Immunol. 2004, 172, 5598–5605. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The Function of Fcγ Receptors in Dendritic Cells and Macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and Immunogenicity of a Recombinant M2e–Flagellin Influenza Vaccine (STF2.4xM2e) in Healthy Adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [CrossRef] [PubMed]
- Talbot, H.K.; Rock, M.T.; Johnson, C.; Tussey, L.; Kavita, U.; Shanker, A.; Shaw, A.R.; Taylor, D.N. Immunopotentiation of Trivalent Influenza Vaccine When Given with VAX102, a Recombinant Influenza M2e Vaccine Fused to the TLR5 Ligand Flagellin. PLoS ONE 2010, 5, e14442. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Osterrieder, N.; Metzger, S.M.; Buckles, E.; Doody, A.M.; DeLisa, M.P.; Putnam, D. Delivery of Foreign Antigens by Engineered Outer Membrane Vesicle Vaccines. Proc. Natl. Acad. Sci. USA 2010, 107, 3099–3104. [Google Scholar] [CrossRef]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune Modulation by Bacterial Outer Membrane Vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef]
- Mamat, U.; Wilke, K.; Bramhill, D.; Schromm, A.B.; Lindner, B.; Kohl, T.A.; Corchero, J.L.; Villaverde, A.; Schaffer, L.; Head, S.R.; et al. Detoxifying Escherichia Coli for Endotoxin-Free Production of Recombinant Proteins. Microb. Cell Fact. 2015, 14, 57. [Google Scholar] [CrossRef]
- Watkins, H.C.; Rappazzo, C.G.; Higgins, J.S.; Sun, X.; Brock, N.; Chau, A.; Misra, A.; Cannizzo, J.P.B.; King, M.R.; Maines, T.R.; et al. Safe Recombinant Outer Membrane Vesicles That Display M2e Elicit Heterologous Influenza Protection. Mol. Ther. 2017, 25, 989–1002. [Google Scholar] [CrossRef]
- Rappazzo, C.G.; Watkins, H.C.; Guarino, C.M.; Chau, A.; Lopez, J.L.; DeLisa, M.P.; Leifer, C.A.; Whittaker, G.R.; Putnam, D. Recombinant M2e Outer Membrane Vesicle Vaccines Protect against Lethal Influenza A Challenge in BALB/c Mice. Vaccine 2016, 34, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Ter Meulen, J.; Casimiro, D.; Coller, B.A.; Heinrichs, J.; Bhambhani, A. Winning a Race against Evolving Pathogens with Novel Platforms and Universal Vaccines; Elsevier Inc.: Amsterdam, The Netherlands, 2015; ISBN 9780124166615. [Google Scholar]
- Hervé, C.; Laupèze, B.; Del Giudice, G.; Didierlaurent, A.M.; Tavares Da Silva, F. The How’s and What’s of Vaccine Reactogenicity. Npj Vaccines 2019, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Belyakov, I.M.; Ahlers, J.D. What Role Does the Route of Immunization Play in the Generation of Protective Immunity against Mucosal Pathogens? J. Immunol. 2009, 183, 6883–6892. [Google Scholar] [CrossRef]
- Anggraeni, R.; Ana, I.D.; Wihadmadyatami, H. Development of Mucosal Vaccine Delivery: An Overview on the Mucosal Vaccines and Their Adjuvants. Clin. Exp. Vaccine Res. 2022, 11, 235–248. [Google Scholar] [CrossRef]
- Stentz, R.; Carvalho, A.L.; Jones, E.J.; Carding, S.R. Fantastic Voyage: The Journey of Intestinal Microbiota-Derived Microvesicles through the Body. Biochem. Soc. Trans. 2018, 46, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.C.; Pagan, C.L.; Childs, H.R.; Posada, S.; Chau, A.; Rios, J.; Guarino, C.; DeLisa, M.P.; Whittaker, G.R.; Putnam, D. A Single Dose and Long Lasting Vaccine against Pandemic Influenza through the Controlled Release of a Heterospecies Tandem M2 Sequence Embedded within Detoxified Bacterial Outer Membrane Vesicles. Vaccine 2017, 35, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Asahi-Ozaki, Y.; Yoshikawa, T.; Iwakura, Y.; Suzuki, Y.; Tamura, S.; Kurata, T.; Sata, T. Secretory IgA Antibodies Provide Cross-Protection against Infection with Different Strains of Influenza B Virus. J. Med. Virol. 2004, 74, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.-I.; Funato, H.; Hirabayashi, Y.; Suzuki, V.; Nagamine, T.; Aizawa, C.; Kurata, T. Cross-Protection against Influenza A Virus Infection by Passively Transferred Respiratory Tract IgA Antibodies to Different Hemagglutinin Molecules. Eur. J. Immunol. 1991, 21, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Mazanec, M.B.; Kaetzel, C.S.; Lamm, M.E.; Fletcher, D.; Nedrud, J.G. Intracellular Neutralization of Virus by Immunoglobulin A Antibodies. Proc. Natl. Acad. Sci. USA 1992, 89, 6901–6905. [Google Scholar] [CrossRef] [PubMed]
- Rajao, D.S.; Zanella, G.C.; Wymore Brand, M.; Khan, S.; Miller, M.E.; Ferreri, L.M.; Caceres, C.J.; Cadernas-Garcia, S.; Souza, C.K.; Anderson, T.K.; et al. Live Attenuated Influenza A Virus Vaccine Expressing an IgA-Inducing Protein Protects Pigs against Replication and Transmission. Front. Virol. 2023, 3, 1042724. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic Identification of Type I and Type II Interferon-Induced Antiviral Factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef]
- Bao, J.; Cui, D.; Wang, X.; Zou, Q.; Zhao, D.; Zheng, S.; Yu, F.; Huang, L.; Dong, Y.; Yang, X.; et al. Decreased Frequencies of Th17 and Tc17 Cells in Patients Infected with Avian Influenza A (H7N9) Virus. J. Immunol. Res. 2019, 2019, 1418251. [Google Scholar] [CrossRef] [PubMed]
- Gnopo, Y.M.D.; Watkins, H.C.; Stevenson, T.C.; DeLisa, M.P.; Putnam, D. Designer Outer Membrane Vesicles as Immunomodulatory Systems–Reprogramming Bacteria for Vaccine Delivery. Adv. Drug Deliv. Rev. 2017, 114, 132–142. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Sanchez, M.V.; Ebensen, T.; Schulze, K.; Cargnelutti, D.; Blazejewska, P.; Scodeller, E.A.; Guzmán, C.A. Intranasal Delivery of Influenza RNP Adjuvanted with C-Di-AMP Induces Strong Humoral and Cellular Immune Responses and Provides Protection against Virus Challenge. PLoS ONE 2014, 9, e104824. [Google Scholar] [CrossRef] [PubMed]
- Prosper, N. Boyaka Inducing Mucosal IgA: A Challenge for Vaccine Adjuvants and Delivery Systems. J. Immunol. Res. 2019, 199, 9–16. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Shu, U.; Kiniwa, M.; Wu, C.Y.; Maliszewski, C.; Vezzio, N.; Hakimi, J.; Gately, M.; Delespesse, G. Activated T Cells Induce Interleukin-12 Production by Monocytes via CD40-CD40 Ligand Interaction. Eur. J. Immunol. 1995, 25, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Stuber, E.; Strober, W.; Neurath, M. Blocking the CD40L-CD40 Interaction in Vivo Specifically Prevents the Priming of T Helper 1 Cells through the Inhibition of Interleukin 12 Secretion. J. Exp. Med. 1996, 183, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Askovich, P.S.; Sanders, C.J.; Rosenberger, C.M.; Diercks, A.H.; Dash, P.; Navarro, G.; Vogel, P.; Doherty, P.C.; Thomas, P.G.; Aderem, A. Differential Host Response, Rather Than Early Viral Replication Efficiency, Correlates with Pathogenicity Caused by Influenza Viruses. PLoS ONE 2013, 8, e74863. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Zhang, X.-E.; Sun, X.; Zhang, X.; Yao, Y.; Liu, S.; Chen, Z.; Li, W.; Zhang, Z.; Chen, J.; et al. Intranasal Nanovaccine Confers Homo- and Hetero-Subtypic Influenza Protection. Small 2018, 14, 1703207. [Google Scholar] [CrossRef]
- Ernst, W.A.; Kim, H.J.; Tumpey, T.M.; Jansen, A.D.A.; Tai, W.; Cramer, D.V.; Adler-Moore, J.P.; Fujii, G. Protection against H1, H5, H6 and H9 Influenza A Infection with Liposomal Matrix 2 Epitope Vaccines. Vaccine 2006, 24, 5158–5168. [Google Scholar] [CrossRef]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding Roles for CD4 + T Cells in Immunity to Viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef]
- Tamura, S.; Kurata, T. Defense Mechanisms against Influenza Virus Infection in the Respiratory Tract Mucosa. Jpn. J. Infect. Dis. 2004, 57, 236–247. [Google Scholar] [CrossRef]
- Hamada, H.; de la L. Garcia-Hernandez, M.; Reome, J.B.; Misra, S.K.; Strutt, T.M.; McKinstry, K.K.; Cooper, A.M.; Swain, S.L.; Dutton, R.W. Tc17, a Unique Subset of CD8 T Cells That Can Protect against Lethal Influenza Challenge. J. Immunol. 2009, 182, 3469–3481. [Google Scholar] [CrossRef] [PubMed]
- Crowe, C.R.; Chen, K.; Pociask, D.A.; Alcorn, J.F.; Krivich, C.; Enelow, R.I.; Ross, T.M.; Witztum, J.L.; Kolls, J.K. Critical Role of IL-17RA in Immunopathology of Influenza Infection. J. Immunol. 2009, 183, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, K.K.; Strutt, T.M.; Buck, A.; Curtis, J.D.; Dibble, J.P.; Huston, G.; Tighe, M.; Hamada, H.; Sell, S.; Dutton, R.W.; et al. IL-10 Deficiency Unleashes an Influenza-Specific Th17 Response and Enhances Survival against High-Dose Challenge. J. Immunol. 2009, 182, 7353–7363. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Yao, X.T.; Peng, Q.; Chen, D.K. The Protective and Pathogenic Roles of IL-17 in Viral Infections: Friend or Foe? Open Biol. 2019, 9, 190109. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of Interleukin 17 Receptor Signaling for Lung Cxc Chemokine and Granulocyte Colony-Stimulating Factor Expression, Neutrophil Recruitment, and Host Defense. J. Exp. Med. 2001, 194, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.D.; Deng, Y.-M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils Ameliorate Lung Injury and the Development of Severe Disease during Influenza Infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef] [PubMed]
- Zalinger, Z.B.; Elliott, R.; Weiss, S.R. Role of the Inflammasome-Related Cytokines Il-1 and Il-18 during Infection with Murine Coronavirus. J. Neurovirol. 2017, 23, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.D.M.; Kenngott, E.E.; Schröter, M.F.; Kühl, A.; Jennrich, S.; Watzlawick, R.; Hoffmann, U.; Wolff, T.; Norley, S.; Scheffold, A.; et al. Timed Action of IL-27 Protects from Immunopathology While Preserving Defense in Influenza. PLoS Pathog. 2014, 10, e1004110. [Google Scholar] [CrossRef] [PubMed]
- Hitchings, M.D.T.; Borgert, B.A.; Shir, A.; Yang, B.; Grantz, K.H.; Ball, J.; Moreno, C.A.; Rand, K.; Small, P.A.; Fowke, K.R.; et al. Dynamics of Anti-Influenza Mucosal IgA Over a Season in a Cohort of Individuals Living or Working in a Long-Term Care Facility. J. Infect. Dis. 2023, 228, 383–390. [Google Scholar] [CrossRef]
- Breedveld, A.; Van Egmond, M. IgA and FcαRI: Pathological Roles and Therapeutic Opportunities. Front. Immunol. 2019, 10, 553. [Google Scholar] [CrossRef]
- Bagheri, Y.; Babaha, F.; Falak, R.; Yazdani, R.; Azizi, G.; Sadri, M.; Abolhassani, H.; Shekarabi, M.; Aghamohammadi, A. IL-10 Induces TGF-β Secretion, TGF-β Receptor II Upregulation, and IgA Secretion in B Cells. Eur. Cytokine Netw. 2019, 30, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Schoenbeck, S.; Mckenzie, D.T.; Kagnoff, M.F. Interleukin 5 Is a Differentiation Factor for IgA B Cells. Eur. J. Immunol. 1989, 19, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.R.; Flannery, B.; Ambrose, C.S.; Bégué, R.E.; Caspard, H.; DeMarcus, L.; Fowlkes, A.L.; Kersellius, G.; Steffens, A.; Fry, A.M. Live Attenuated and Inactivated Influenza Vaccine Effectiveness. Pediatrics 2019, 143, e20182094. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kannan, N.; Choi, A.; Rivera De Jesus, M.A.; Wei, P.M.; Sahler, J.M.; Curley, S.M.; August, A.; DeLisa, M.P.; Whittaker, G.R.; Putnam, D. Intranasal Vaccination with Recombinant TLR2-Active Outer Membrane Vesicles Containing Sequential M2e Epitopes Protects against Lethal Influenza a Challenge. Vaccines 2024, 12, 724. https://doi.org/10.3390/vaccines12070724
Kannan N, Choi A, Rivera De Jesus MA, Wei PM, Sahler JM, Curley SM, August A, DeLisa MP, Whittaker GR, Putnam D. Intranasal Vaccination with Recombinant TLR2-Active Outer Membrane Vesicles Containing Sequential M2e Epitopes Protects against Lethal Influenza a Challenge. Vaccines. 2024; 12(7):724. https://doi.org/10.3390/vaccines12070724
Chicago/Turabian StyleKannan, Nisha, Annette Choi, Mariela A. Rivera De Jesus, Peter Male Wei, Julie Marie Sahler, Stephanie Marie Curley, Avery August, Matthew P. DeLisa, Gary R. Whittaker, and David Putnam. 2024. "Intranasal Vaccination with Recombinant TLR2-Active Outer Membrane Vesicles Containing Sequential M2e Epitopes Protects against Lethal Influenza a Challenge" Vaccines 12, no. 7: 724. https://doi.org/10.3390/vaccines12070724