
Therapeutic Anti-Tumor Efficacy of DC-Based Vaccines Targeting TME-Associated Antigens Is Improved When Combined with a Chemokine-Modulating Regimen and/or Anti-PD-L1

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
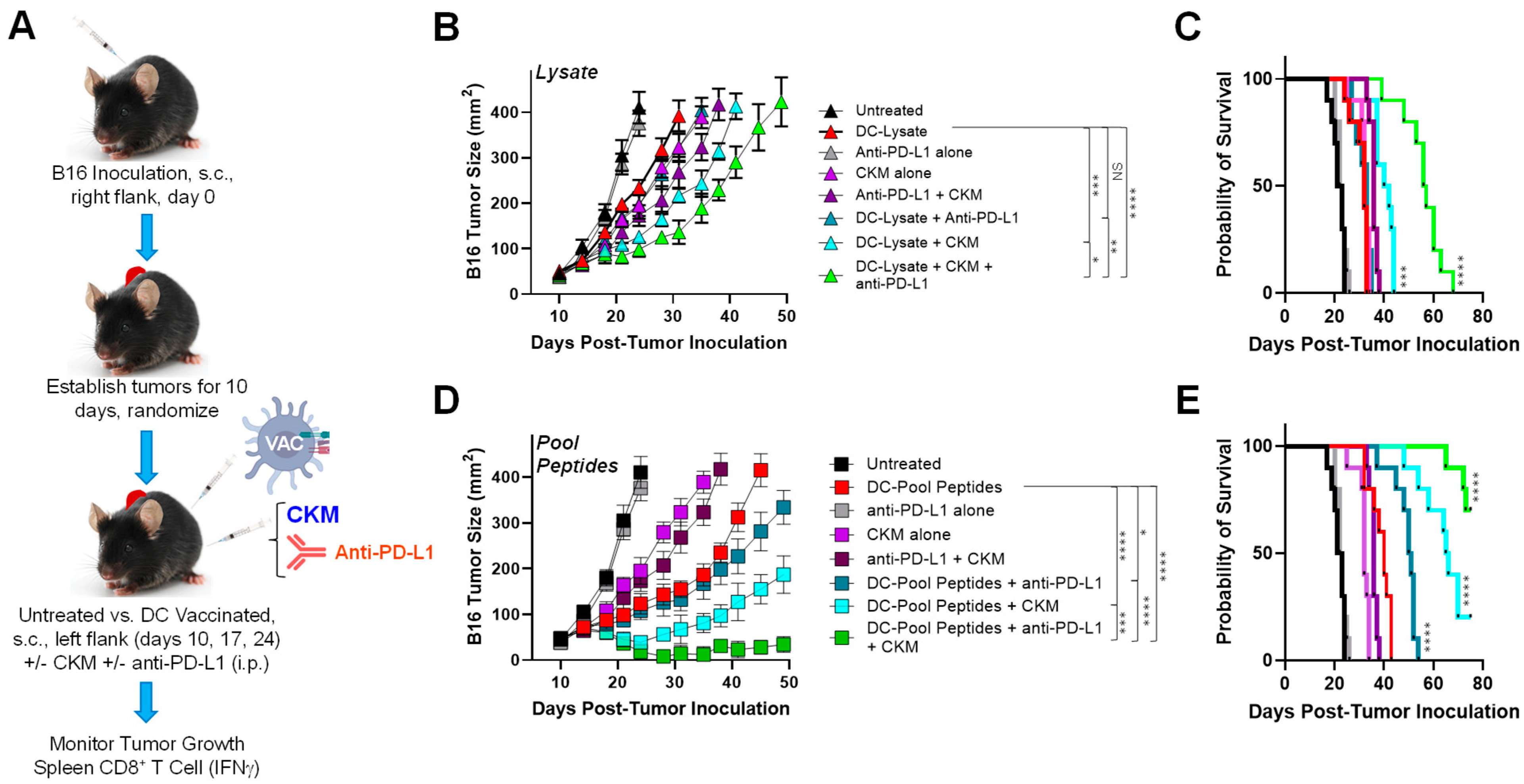
3.1. Coordinate Vaccine Targeting of Melanoma and Tumor-Associated Stromal Antigens Effectively Controls Melanoma Growth In Vivo
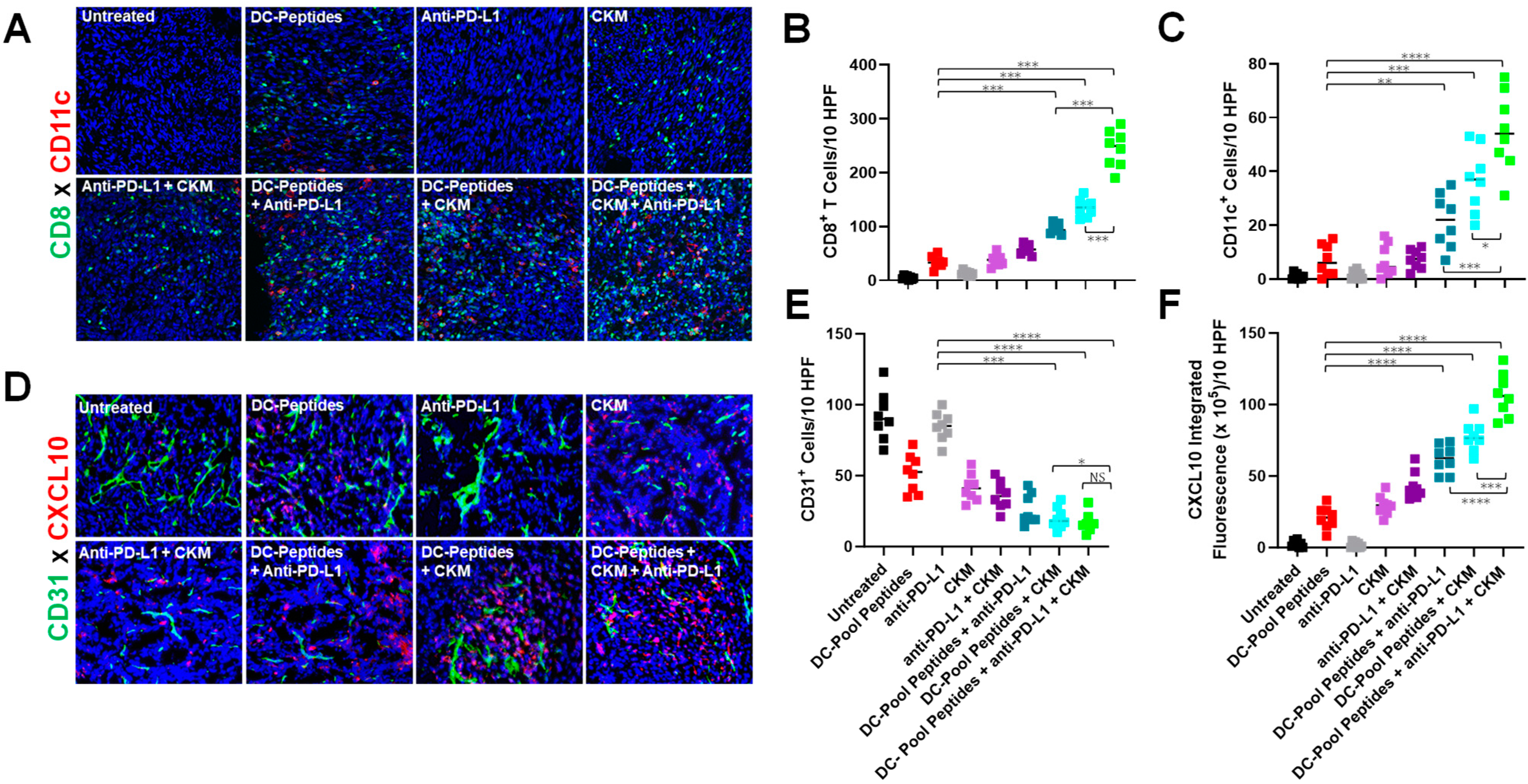
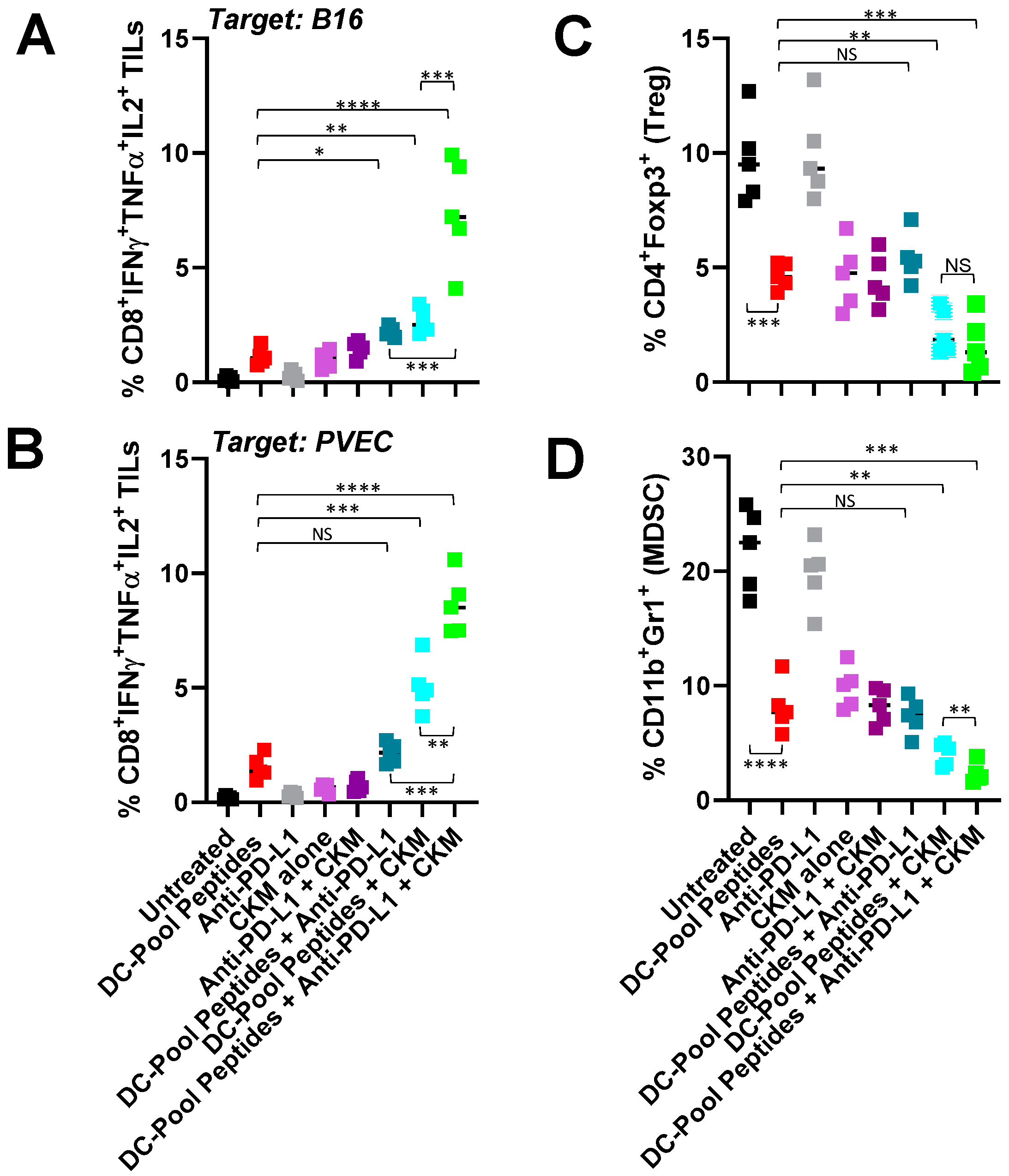
3.2. Immunogenic and Immunomodulatory Activity of DC-Based Vaccines Combined with CKM and/or Anti-PD-L1 Blockade
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. Cancer vaccines. Immunol. Today 1993, 14, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.I.; Huis In ‘t Veld, L.G.M.; Raaijmakers, T.K.; Adema, G.J. Adjuvants Enhancing Cross-Presentation by Dendritic Cells: The Key to More Effective Vaccines? Front. Immunol. 2018, 9, 2874. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.; Salomon, N.; Wolf, Y.; Kreiter, S.; Diken, M.; Samuels, Y. The landscape of T cell antigens for cancer immunotherapy. Nat. Cancer 2023, 4, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. Tumor microenvironment antigens. Semin. Immunopathol. 2023, 45, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bose, A.; Komita, H.; Taylor, J.L.; Chi, N.; Lowe, D.B.; Okada, H.; Cao, Y.; Mukhopadhyay, D.; Cohen, P.A.; et al. Vaccines targeting tumor blood vessel antigens promote CD8+ T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. J. Immunol. 2012, 188, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Chi Sabins, N.; Taylor, J.L.; Fabian, K.P.; Appleman, L.J.; Maranchie, J.K.; Stolz, D.B.; Storkus, W.J. DLK1: A novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol. Ther. 2013, 21, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Pan, Z.K.; Guirnalda, P.; Tsai, P.; Seavey, M.; Paterson, Y. Targeting tumor vasculature with novel Listeria-based vaccines directed against CD105. Cancer Immunol. Immunother. 2011, 60, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, A.G.; Xiang, R.; Becker, J.C.; Wodrich, H.; Pertl, U.; Karsten, G.; Eliceiri, B.P.; Reisfeld, R.A. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat. Med. 2002, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Storkus, W.J.; Maurer, D.; Lin, Y.; Ding, F.; Bose, A.; Lowe, D.; Rose, A.; DeMark, M.; Karapetyan, L.; Taylor, J.L.; et al. Dendritic cell vaccines targeting tumor blood vessel antigens in combination with dasatinib induce therapeutic immune responses in patients with checkpoint-refractory advanced melanoma. J. Immunother. Cancer 2021, 9, e003675. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018, 39, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Moi, D.; Zeng, B.; Minnie, S.A.; Bhatt, R.; Wood, J.; Sester, D.P.; Mazzieri, R.; Dolcetti, R. Multiparametric flow cytometry to characterize vaccine-induced polyfunctional T cell responses and T cell/NK cell exhaustion and memory phenotypes in mouse immuno-oncology models. Front. Immunol. 2023, 14, 1127896. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Urban, J.; Wieckowski, E.; Muthuswamy, R.; Ravindranathan, R.; Bartlett, D.L.; Kalinski, P. Promoting the accumulation of tumor-specific T cells in tumor tissues by dendritic cell vaccines and chemokine-modulating agents. Nat. Protoc. 2018, 13, 335–357. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Mackay, A.; Vohra, N.; Mulé, J.J. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J. Immunol. 2010, 184, 3442–3449. [Google Scholar] [CrossRef] [PubMed]
- Overwijk, W.W.; Tsung, A.; Irvine, K.R.; Parkhurst, M.R.; Goletz, T.J.; Tsung, K.; Carroll, M.W.; Liu, C.; Moss, B.; Rosenberg, S.A.; et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998, 188, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Patiño, J.A.; Engelhorn, M.E.; Turk, M.J.; Liu, C.; Duan, F.; Rizzuto, G.; Cohen, A.D.; Merghoub, T.; Wolchok, J.D.; Houghton, A.N. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J. Clin. Investig. 2006, 116, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- van Elsas, A.; Sutmuller, R.P.; Hurwitz, A.A.; Ziskin, J.; Villasenor, J.; Medema, J.P.; Overwijk, W.W.; Restifo, N.P.; Melief, C.J.; Offringa, R.; et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: Comparison of prophylaxis and therapy. J. Exp. Med. 2001, 194, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Kuwashima, N.; Tatsumi, T.; Dusak, J.E.; Nishimura, F.; Reilly, K.M.; Storkus, W.J.; Okada, H. Vaccination with EphA2-derived T cell-epitopes promotes immunity against both EphA2-expressing and EphA2-negative tumors. J. Transl. Med. 2004, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Feltkamp, M.C.; Smits, H.L.; Vierboom, M.P.; Minnaar, M.P.; de Jongh, B.M.; Drijfhout, J.W.; ter Schegget, J.; Melief, C.J.; Kast, W.M. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol. 1993, 23, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Herr, W.; Ranieri, E.; Olson, W.; Zarour, H.; Gesualdo, L.; Storkus, W.J. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4+ and CD8+ T lymphocyte responses. Blood 2000, 96, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Carey, J.B.; Li, Y.; Spencer, A.J.; van Laarhoven, A.; Mullarkey, C.E.; Vrdoljak, A.; Moore, A.C.; Gilbert, S.C. Immunity against heterosubtypic influenza virus induced by adenovirus and MVA expressing nucleoprotein and matrix protein-1. Sci. Rep. 2013, 3, 1443. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Woods, A.; Fu, T.; He, Q.; Nielsen, M.C.; Hasan, F.; Roszik, J.; Xiao, Z.; Vianden, C.; Khong, H.; et al. Cancer vaccine formulation dictates synergy with CTLA-4 and PD-L1 checkpoint blockade therapy. J. Clin. Investig. 2018, 128, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Thomas, E.K.; Giedlin, M.; Mulé, J.J. Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res. 2001, 61, 2618–2624. [Google Scholar]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin. Cancer Res. 2020, 26, 4442–4447. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Timmerman, J.M.; Butterfield, L.H.; Economou, J.S. Determinant spreading and tumor responses after peptide-based cancer immunotherapy. Trends Immunol. 2003, 24, 58–61. [Google Scholar] [CrossRef] [PubMed]
- el-Shami, K.; Tirosh, B.; Bar-Haim, E.; Carmon, L.; Vadai, E.; Fridkin, M.; Feldman, M.; Eisenbach, L. MHC class I-restricted epitope spreading in the context of tumor rejection following vaccination with a single immunodominant CTL epitope. Eur. J. Immunol. 1999, 29, 3295–3301. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Vercher, E.; Soria-Castellano, M.; Suarez-Olmos, J.; Mancheño, U.; Elizalde, E.; Rodriguez, M.L.; Glez-Vaz, J.; Casares, N.; Rodríguez-García, E.; et al. Epitope spreading driven by the joint action of CART cells and pharmacological STING stimulation counteracts tumor escape via antigen-loss variants. J. Immunother. Cancer 2021, 9, e003351. [Google Scholar] [CrossRef] [PubMed]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signaling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; Gawrońska-Szklarz, B.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors-A Review of Literature. Int. J. Mol. Sci. 2021, 22, 843. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy-current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.M.; Titmuss, E.; Wang, Y.; Vafiadis, J.; Pacis, A.; Jang, G.H.; Zhang, A.; Golesworthy, B.; Lenko, T.; Williamson, L.M.; et al. Chemokine expression predicts T cell-inflammation and improved survival with checkpoint inhibition across solid cancers. NPJ Precis. Oncol. 2023, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Shan, F.; Somasundaram, A.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer 2022, 8, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.T.; Gabrilovich, D.I.; Sansom, O.J.; Campbell, A.D.; Morton, J.P. Therapeutic targeting of tumour myeloid cells. Nat. Rev. Cancer 2023, 23, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancer-associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549. [Google Scholar] [CrossRef] [PubMed]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef] [PubMed]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Yu, F.; Ji, M.; Kakarla, S.; Song, X.T. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS ONE 2013, 8, e82658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Duan, X.; Liu, Y.; Xu, J.; Al-Bashari, A.A.G.; Ye, P.; Ye, Q.; He, Y. The Application of Mesenchymal Stem Cells in Future Vaccine Synthesis. Vaccines 2023, 11, 1631. [Google Scholar] [CrossRef] [PubMed]
- Tomchuck, S.L.; Norton, E.B.; Garry, R.F.; Bunnell, B.A.; Morris, C.A.; Freytag, L.C.; Clements, J.D. Mesenchymal stem cells as a novel vaccine platform. Front. Cell Infect. Microbiol. 2012, 2, 140. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, J.L.; Kokolus, K.M.; Basse, P.H.; Filderman, J.N.; Cosgrove, C.E.; Watkins, S.C.; Gambotto, A.; Lowe, D.B.; Edwards, R.P.; Kalinski, P.; et al. Therapeutic Anti-Tumor Efficacy of DC-Based Vaccines Targeting TME-Associated Antigens Is Improved When Combined with a Chemokine-Modulating Regimen and/or Anti-PD-L1. Vaccines 2024, 12, 777. https://doi.org/10.3390/vaccines12070777
Taylor JL, Kokolus KM, Basse PH, Filderman JN, Cosgrove CE, Watkins SC, Gambotto A, Lowe DB, Edwards RP, Kalinski P, et al. Therapeutic Anti-Tumor Efficacy of DC-Based Vaccines Targeting TME-Associated Antigens Is Improved When Combined with a Chemokine-Modulating Regimen and/or Anti-PD-L1. Vaccines. 2024; 12(7):777. https://doi.org/10.3390/vaccines12070777
Chicago/Turabian StyleTaylor, Jennifer L., Kathleen M. Kokolus, Per H. Basse, Jessica N. Filderman, Chloe E. Cosgrove, Simon C. Watkins, Andrea Gambotto, Devin B. Lowe, Robert P. Edwards, Pawel Kalinski, and et al. 2024. "Therapeutic Anti-Tumor Efficacy of DC-Based Vaccines Targeting TME-Associated Antigens Is Improved When Combined with a Chemokine-Modulating Regimen and/or Anti-PD-L1" Vaccines 12, no. 7: 777. https://doi.org/10.3390/vaccines12070777