
Lessons from Post-Immunotherapy Tumor Tissues in Clinical Trials: How Can We Fuel the Tumor Microenvironment in Gliomas?


Abstract
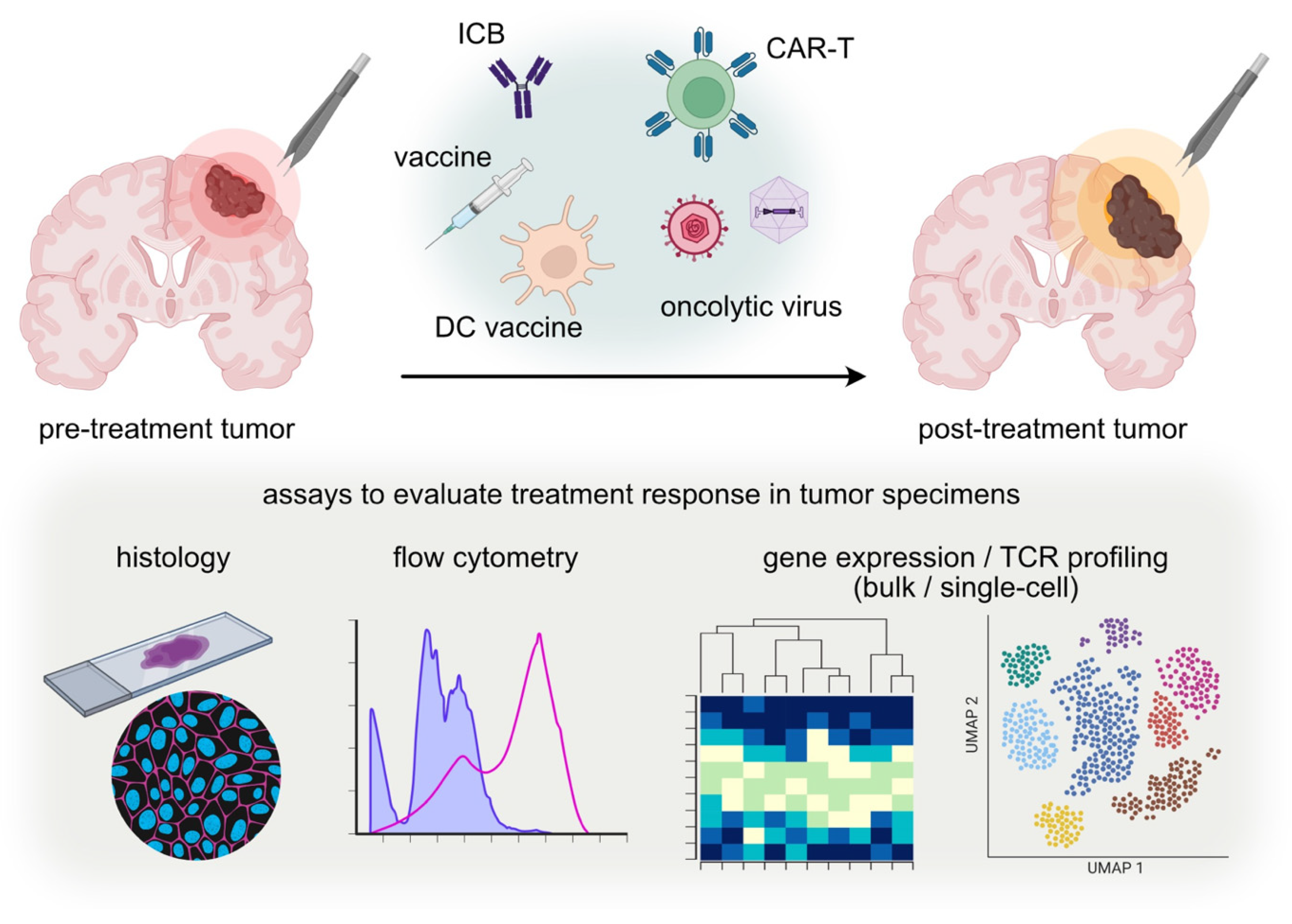
:1. Introduction
2. Methodology
2.1. Analysis Data of Post-Treatment Tumor Tissue at Disease Progression
2.2. Analysis Data of Post-Treatment Tumor Tissues at a Predefined Timing
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Haslam, A.; Gill, J.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Are Eligible for Immune Checkpoint Inhibitor Drugs. JAMA Netw. Open 2020, 3, e200423. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chuntova, P.; Chow, F.; Watchmaker, P.B.; Galvez, M.; Heimberger, A.B.; Newell, E.W.; Diaz, A.; Depinho, R.A.; Li, M.O.; Wherry, E.J. Unique Challenges for Glioblastoma Immunotherapy-Discussions across Neuro-Oncology and Non-Neuro-Oncology Experts in Cancer Immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro. Oncol. 2021, 23, 356–375. [Google Scholar]
- Weller, M.; Roth, P.; Preusser, M.; Wick, W.; Reardon, D.A.; Platten, M.; Sampson, J.H. Vaccine-Based Immunotherapeutic Approaches to Gliomas and Beyond. Nat. Rev. Neurol. 2017, 13, 363–374. [Google Scholar] [CrossRef]
- Buerki, R.A.; Chheda, Z.S.; Okada, H. Immunotherapy of Primary Brain Tumors: Facts and Hopes. Clin. Cancer Res. 2018, 24, 5198–5205. [Google Scholar]
- Bagley, S.J.; Kothari, S.; Rahman, R.; Lee, E.Q.; Dunn, G.P.; Galanis, E.; Chang, S.M.; Nabors, L.B.; Ahluwalia, M.S.; Stupp, R.; et al. Glioblastoma Clinical Trials: Current Landscape and Opportunities for Improvement. Clin. Cancer Res. 2022, 28, 594–602. [Google Scholar] [CrossRef]
- Burster, T.; Traut, R.; Yermekkyzy, Z.; Mayer, K.; Westhoff, M.-A.; Bischof, J.; Knippschild, U. Critical View of Novel Treatment Strategies for Glioblastoma: Failure and Success of Resistance Mechanisms by Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 695325. [Google Scholar] [CrossRef] [PubMed]
- Levit, L.A.; Peppercorn, J.M.; Tam, A.L.; Marron, J.M.; Mathews, D.J.H.; Levit, K.; Roach, N.; Ratain, M.J. Ethical Framework for Including Research Biopsies in Oncology Clinical Trials: American Society of Clinical Oncology Research Statement. J. Clin. Oncol. 2019, 37, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable Interleukin-12 Gene Therapy in Patients with Recurrent High-Grade Glioma: Results of a Phase 1 Trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M. Phase I/II Trial Testing Safety and Immunogenicity of the Multipeptide IMA950/Poly-ICLC Vaccine in Newly Diagnosed Adult Malignant Astrocytoma Patients. Neuro. Oncol. 2019, 21, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D. Combined Immunotherapy with Controlled Interleukin-12 Gene Therapy and Immune Checkpoint Blockade in Recurrent Glioblastoma: An Open-Label, Multi-Institutional Phase I Trial. Neuro. Oncol. 2021, 24, 951–996. [Google Scholar] [CrossRef]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral Administration of CTLA-4 and PD-1 Immune Checkpoint Blocking Monoclonal Antibodies in Patients with Recurrent Glioblastoma: A Phase I Clinical Trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-Shelf, Steroid-Resistant, IL13Rα2-Specific CAR T Cells for Treatment of Glioblastoma. Neuro. Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Gállego Pérez-Larraya, J.; Garcia-Moure, J.; Labiano, M.; Patiño-García, S.; Dobbs, A.; Gonzalez-Huarriz, J.; Zalacain, M.; Marrodan, M.; Martinez-Velez, L.; Puigdelloses, N. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical Trial Links Oncolytic Immunoactivation to Survival in Glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.-Y.; Chen, T.; Chang, L.-J.; et al. Safety and Antitumor Activity of GD2-Specific 4SCAR-T Cells in Patients with Glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.P.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G. Repeated Peripheral Infusions of Anti-EGFRvIII CAR T Cells in Combination with Pembrolizumab Show No Efficacy in Glioblastoma: A Phase 1 Trial. Nat. Cancer 2024, 5, 517–553. [Google Scholar] [PubMed]
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–525. [Google Scholar] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; López-Diaz De Cerio, A.; Tejada, S. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A Phase I/II Study of Triple-Mutated Oncolytic Herpes Virus G47∆ in Patients with Progressive Glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef]
- Kasenda, B.; König, D.; Manni, M.; Ritschard, R.; Duthaler, U.; Bartoszek, E.; Bärenwaldt, A.; Deuster, S.; Hutter, G.; Cordier, D. Targeting Immunoliposomes to EGFR-Positive Glioblastoma. ESMO Open 2022, 7, 100365. [Google Scholar] [CrossRef]
- Ogino, H.; Taylor, J.W.; Nejo, T.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Saijo, A.; Tedesco, M.R.; Shai, A.; Wong, C.M.; et al. Randomized Trial of Neoadjuvant Vaccination with Tumor-Cell Lysate Induces T Cell Response in Low-Grade Gliomas. J. Clin. Investig. 2022, 132, e151239. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Saijo, A.; Ogino, H.; Butowski, N.A.; Tedesco, M.R.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Wang, A.S.; Shai, A.; Salazar, A.M.; et al. A Combinatory Vaccine with IMA950 plus Varlilumab Promotes Effector Memory T-Cell Differentiation in the Peripheral Blood of Patients with Low-Grade Gliomas. Neuro. Oncol. 2024, 26, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Dooley, K.E.; Keith Anderson, S.; Kurokawa, C.B.; Carrero, X.W.; Uhm, J.H.; Federspiel, M.J.; Leontovich, A.A.; Aderca, I.; Viker, K.B. Carcinoembryonic Antigen-Expressing Oncolytic Measles Virus Derivative in Recurrent Glioblastoma: A Phase 1 Trial. Nat. Commun. 2024, 15, 493. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Maclean, P.; Nasrallah, A.; Desai, J.J.; Melenhorst, K.; Mansfield, J.J.D.; Morrissette, M. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; van Zwieten, K.; Prince, J.; van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M.; et al. Changes in the EGFR Amplification and EGFRvIII Expression between Paired Primary and Recurrent Glioblastomas. Neuro. Oncol. 2015, 17, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ Regulatory T Cells and Their Functional Regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A. How Do Regulatory T Cells Work? Scand. Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hosseinalizadeh, H.; Rabiee, F.; Eghbalifard, N.; Rajabi, H.; Klionsky, D.J.; Rezaee, A. Regulating the Regulatory T Cells as Cell Therapies in Autoimmunity and Cancer. Front. Med. (Lausanne) 2023, 10, 1244298. [Google Scholar] [CrossRef]
- Weathers, S.-P.; Penas-Prado, M.; Pei, B.-L.; Ling, X.; Kassab, C.; Banerjee, P.; Bdiwi, M.; Shaim, H.; Alsuliman, A.; Shanley, M. Glioblastoma-Mediated Immune Dysfunction Limits CMV-Specific T Cells and Therapeutic Responses: Results from a Phase I/II Trial. Clin. Cancer Res. 2020, 26, 3565–3577. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. Author Correction: EANO Guidelines on the Diagnosis and Treatment of Diffuse Gliomas of Adulthood. Nat. Rev. Clin. Oncol. 2022, 19, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Anti-EGFRvIII SynNotch Receptor Induced Anti-EphA2/IL-13Ralpha2 CAR (E-SYNC) T Cells. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT06186401 (accessed on 24 June 2024).
- Karschnia, P.; Smits, M.; Reifenberger, G.; Le Rhun, E.; Ellingson, B.M.; Galldiks, N.; Kim, M.M.; Huse, J.T.; Schnell, O.; Harter, P.N. A Framework for Standardised Tissue Sampling and Processing during Resection of Diffuse Intracranial Glioma: Joint Recommendations from four RANO groups. Lancet. Oncol. 2023, 24, e438–e450. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Author, Year | Trial Registration | Disease | Treatment Investigated | n (Patients) | Key Assays and Findings |
|---|---|---|---|---|---|
| Chiocca EA, 2019 [13] | NCT02026271 (ATI001-102) | Rec-GBM or anaplastic astro (grade 3) | Regulatable interleukin-12 gene therapy (tumor cavity injection) | 5 | 4 out of 5 patients were found to be pseudoprogression. IHC/IF: increased tumor-infiltrating lymphocytes producing IFNγ and PD-1. Intratumoral IFNγ concentration increased after gene therapy. |
| Hilf N, 2019 [14] | NCT02149225 (GAPVAC-101) | ND-GBM | Personalized peptide vaccines (unmutated, or unmutated and mutated) | 1 | IHC: high infiltration by T cells with a favorable CD8+ T/FOXP3+ Treg cell ratio. CD4+ T-cell response: the tumor contained CD4+ T cells directed against the APVAC1 pan-DR peptide PTP-010. |
| Keskin, 2019 [15] | NCT02287428 (NeoVax trial) | MGMT-unmethylated, ND-GBM | Personalized neoantigen vaccines | 5 | Multiplexed IF: a significant increase in infiltrating CD8+ T cells at relapse in 2 patients, whereas no increase was observed in the other 3 patients who received dexamethasone. TCR repertoire seq: a subset of putatively reactive TCR α and β-chain sequences were directly detectable in the post-treatment sample RNA, suggesting the successful trafficking to the site of disease. scRNA-seq: nearly all CD8+ T cells appeared to be differentiated cells (CCR7–) and expressed markers of cytotoxicity (PRF1, GZMA, and GZMK). |
| Migliorini D, 2019 [16] | NCT01920191 | ND-GBM and grade 3 astrocytoma | IMA950 multi-peptide vaccine and poly-ICLC | 7 | IHC: no major changes in antigen expression were observed in recurrent samples. No correlation was observed between tumor antigen expression and antigen-specific CD8+ T-cell responses. |
| Chiocca EA, 2021 [17] | NCT03636477 | Rec-GBM | Ad-RTS-hIL-12: Veledimex (VDX)-regulatable IL-12 gene therapy with neoadjuvant nivolumab (anti-PD-1 Ab) | 4 | LC-MS: a dose–response relationship with effective brain tumor tissue VDX penetration was observed. Histology/multiplexed-IF: a significant decrease in the number of PD-1+ cells and PD-L1+ cells was observed. The addition of ICBs reduced PD-1/PD-L1 expression. Activated TILs also decreased between pre-and post-treatment tissues. |
| Duerinck J, 2021 [18] | NCT03233152 | Rec-GBM | Preoperative nivolumab and peritumoral administration of nivo or nivolumab + ipilimumab | 3 | Histology: no evidence of tumor recurrence, but immune cell infiltration was observed in 2 out of 3 tumors. |
| Friedman GK, 2021 [19] | NCT02457845 | pediatric HGG | G207: Oncolytic HSV-1 | 4 | IHC: HSV-1 staining was completely negative in any post-treatment tissues, which indicated that G207 was no longer replicating. A brisk infiltration of CD8+ T cells and increases in CD20+ B-cells and CD138+ plasma cells were revealed. |
| Platten M, 2021 [20] | NCT02454634 (NOA16) | ND-, WHO grades 3 and 4 IDH1(R132H)+ astrocytoma | IDH1-vac: IDH1(R132H)-specific peptide vaccine | 1 | ELISPOT: IDH1(R132H)-reactive T cells were identified from lesion-infiltrating leukocytes (LILs). scRNA-seq/scTCR-seq: among the three clusters of CD4+ T cells, two non-regulatory T-cell clusters were dominated by one TCR (“TCR14”). TCR14 was enriched 50.6-fold in the PsPD lesion compared to the patient’s PBMC. |
| Brown CE, 2022 [21] | NCT01082926 | Rec-GBM | GRm13Z40-2 cells: healthy-donor-derived IL13Rα2-targeted CAR T cell | 2 | IHC: IL13Rα2 expression was maintained, and CD8+ T-cell infiltration increased. FISH: only limited numbers of GRm13Z40-2 cells persisted since treatment. |
| Gállego Pérez-Larraya J, 2022 [22] | NCT03178032 | Pediatric, ND-DIPG | DNX-2401: an oncolytic adenovirus | 1 | Multiplex IF: at relapse, increases in CD8+ and CD4+ T cells and a decrease in myeloid cells were observed. In contrast, reductions in CD8+ and CD4+ T cells and an increase in CD163+ M2 macrophages were observed. sn-RNA-seq: after treatment, tumor-infiltrating macrophages showed upregulations of viral process and immune response pathways. |
| Ling AL, 2023 [23] | NCT03152318 | Rec-GBM | CAN-3110: an oncolytic herpes virus (oHSV) | 29 | PCR: the presence of CAN-3110-specific viral DNA was confirmed. Histology/IHC: increases in CD8+ and CD4+ TILs. TCRβ-DNA-seq: increased tumor TCRβ diversity was associated with prolonged post-treatment survival. RNA-seq: a highly inflammatory and immunologically activated TME in HSV1 serologically positive patients. |
| Liu Z, 2023 [24] | NCT03170141 | GD2+, Rec-or progressive GBM | GD2-specific 4S-CAR T cells | 1 | IHC/IF: GD2 antigen loss and T-cell infiltration were observed. |
| Bagley SJ, 2024 [25] | NCT03726515 | EGFRvIII+, ND-GBM | Anti-EGFRvIII-CAR T with pembrolizumab (anti-PD-1 Ab). | 7 | qPCR: only in 1 out of 7 tumors and CAR T cells were detected in the brain via BBZ qPCR. scRNA-seq: no CAR T cells were found, including in the qPCR-positive case. Increases in exhaustion markers and IFN-stimulated signatures were observed after treatment. |
| Choi BD, 2024 [26] | NCT05660369 (INCIPIENT study) | EGFRvIII+, ND- or Rec-GBM | CARv3-TEAM-E T cells: EGFRvIII-CAR also secreting T-cell-engaging antibody molecules [TEAM] against wt-EGFR | 1 | NGS and IHC: negative for EGFRvIII, while a gain in the EGFR copy number was maintained. |
| Author, Year | Trial Registration | Disease | Treatment Investigated | n (Patients) | Time from Treatment | Key Assays and Findings |
|---|---|---|---|---|---|---|
| Cloughesy TF, 2019 [27] | N/A | Rec-GBM | Neoadjuvant anti-PD-1 (pembrolizumab) (vs. adjuvant only) | 14 | 14 days +/− 5 | RNA-seq: upregulation of T-cell- and interferon-γ-related gene expressions, but downregulation of cell-cycle-related gene expression within the tumor. IF: neoadjuvant anti-PD-1 treatment is associated with focal induction of PD-L1 expression with a high CD8 infiltrate. TCR-seq: neoadjuvant anti-PD-1 uniquely initiated a coordinated local and systemic T-cell response. |
| Schalper KA, 2019 [28] | NCT02550249 | ND- and rec-GBM | Neoadjuvant nivolumab (anti-PD-1 Ab) | 30 | 14 days +/− 3 | Nanostring: Nivo-treated samples showed an upregulation of numerous immune-related transcripts. FCM: most CD8+ eff cells expressed CD69 and HLA-DR, indicating activation and/or tissue residence. IHC (in 3 cases): confirmed at least partial receptor occupancy at the time of surgery, as revealed by differential staining treatments using mAbs targeting PD-1 (in 3 cases). TCR-seq: increased clonal T-cell diversity following neoadjuvant Nivo treatment. Multiplexed IF: Nivo treatment was associated with a minimal change or a mild increase in immune cell markers, whereas the standard treatment (control) was associated with a global reduction in both adaptive and innate immune cell indicators. |
| Weathers SP, 2020 [29] | NCT02661282 | ND- and rec-GBM | Autologous CMV pp65-specific T cells | 1 | 8 days | ELISA: CD8+ T cells isolated from GBM-TME were more refractory to stimulation and unreactive to CMV-peptide stimulation. IHC: CMV-specific CD8+ T cells were PD-1 positive, mostly in the tumor vasculature and not spreading, indicating they were dysfunctional. |
| Kasenda B, 2022 [30] | NCT03603379 (GBM-LIPO trial) | EGFR-amplified, Rec-GBM | Anti-EGFR ILs-dox: anti-EGFR immunoliposomes loaded with doxorubicin | 3 | 24 h | PK: doxorubicin was detectable in the tumor tissues 24 h after treatment, whereas it was undetectable in CSF. IHC/IMC: CD68+ macrophage population was relatively more frequent in two patients after treatment, while a clear reduction, along with a lower proliferation of glioma cells, was observed in the other patient. |
| Ogino H, 2022 [31] | NCT02549833 | ND- or rec-WHO grade 2 gliomas | GBM6-AD: allogeneic cell lysate-based vaccine | 13 | 2 days after 4 cycles of vaccines | TCRβ-seq: some TCRβ clonotypes enriched in post-vaccinated peripheral blood were also identified in the corresponding tumor tissue, suggesting the successful migration of the vaccine-reactive T cells into the TME. Mass cytometry: the proportion of CD103+CD8+ T cells with an effector memory phenotype was significantly higher in tumors in the neoadjuvant vaccine group, with a higher positivity for CXCR3. |
| Todo T, 2022 [29] | UMIN000002661 | Rec- or progressive GBM | G47∆: a triple-mutated oncolytic HSV type 1 | 13 | 0 | IHC: decreased number of tumor cells, infiltration of CD4+ and CD8+ T cells, and HSV-1 positive staining were observed. |
| Todo T, 2022 [32] | UMIN000015995 | Rec- or residual GBM | G47∆: a triple-mutated oncolytic HSV type 1 | 19 (3) | 0 | IHC: it was confirmed that all recurrent cases were not pseudoprogression. Increased numbers of CD4+ and CD8+ T-cell infiltration and persistent low numbers of Foxp3+ cells were observed. At tumor progression, increased numbers of Foxp3+ cells were found in the two cases at 4 months. |
| Saijo A, 2023 [33] | NCT02924038 | WHO grade 2 LGG | IMA950 multi-peptide vaccine ± varlilumab (agonistic anti-CD27 Ab) | 10 | 2 days after 4 cycles of vaccines | Mass cytometry: adding varlilumab induced detectable changes in PBMCs but not in the TME. |
| Galanis E, 2024 [34] | NCT00390299 | Rec-GBM | CEA-MV: CEA-expressing oncolytic measles virus derivative | 11 (5) | 5 days | Nanostring: the gene expression scores of interferon-stimulated genes were inversely correlated with virus replication. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phung, L.H.; Nejo, T.; Okada, H. Lessons from Post-Immunotherapy Tumor Tissues in Clinical Trials: How Can We Fuel the Tumor Microenvironment in Gliomas? Vaccines 2024, 12, 862. https://doi.org/10.3390/vaccines12080862
Phung LH, Nejo T, Okada H. Lessons from Post-Immunotherapy Tumor Tissues in Clinical Trials: How Can We Fuel the Tumor Microenvironment in Gliomas? Vaccines. 2024; 12(8):862. https://doi.org/10.3390/vaccines12080862
Chicago/Turabian StylePhung, Lan Hoc, Takahide Nejo, and Hideho Okada. 2024. "Lessons from Post-Immunotherapy Tumor Tissues in Clinical Trials: How Can We Fuel the Tumor Microenvironment in Gliomas?" Vaccines 12, no. 8: 862. https://doi.org/10.3390/vaccines12080862