
Characterization of the Anti-Viral and Vaccine-Specific CD8+ T Cell Composition upon Treatment with the Cancer Vaccine VSV-GP

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Tumor Cell Line and Tumor Cell Implantation
2.3. Generation of Vaccine Constructs and Immunizations
2.4. Tissue Harvest
2.5. Flow Cytometry
2.6. Data Analysis and Statistics
3. Results
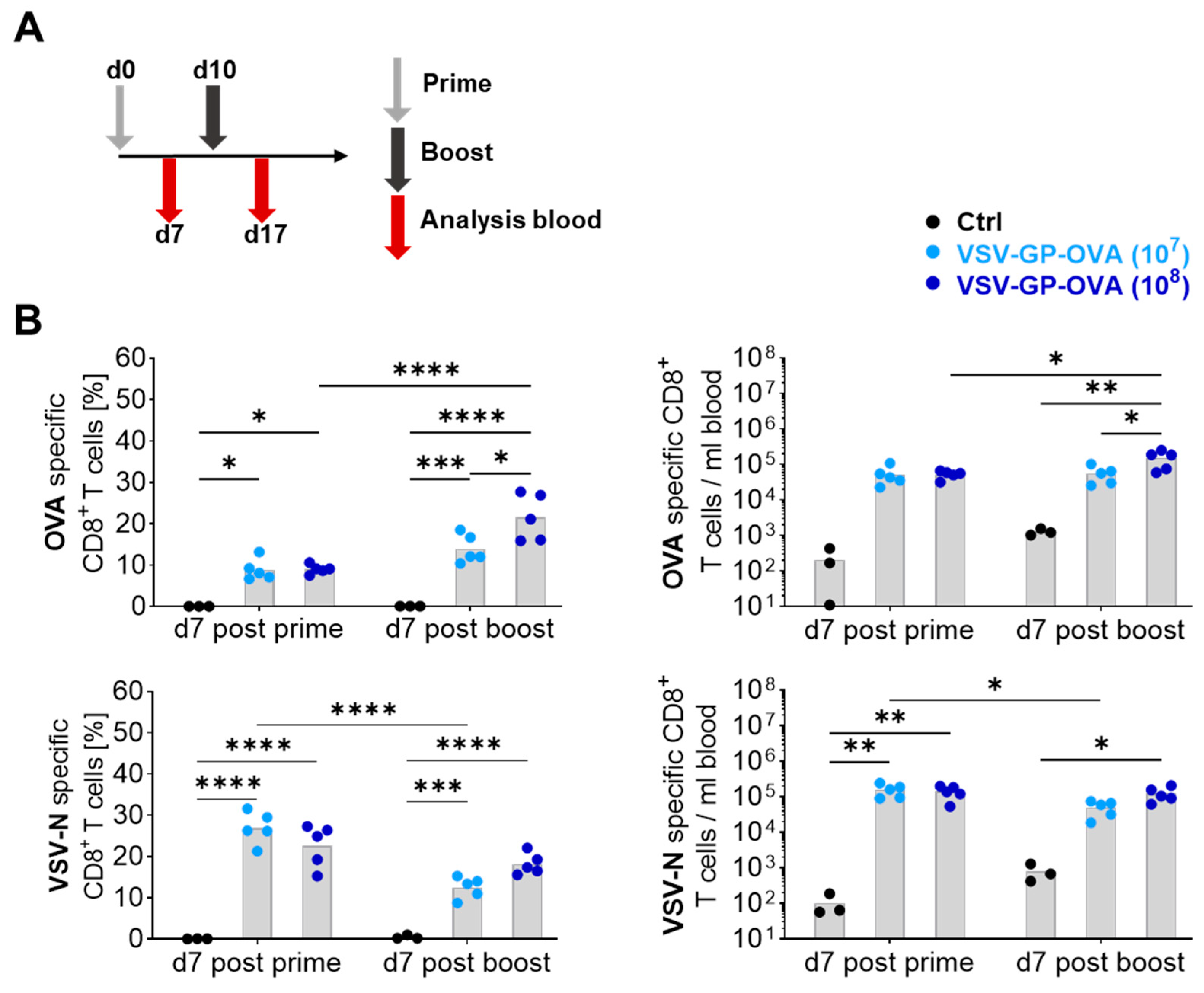
3.1. Vaccine Dose Influences the CD8+ T Cell Response
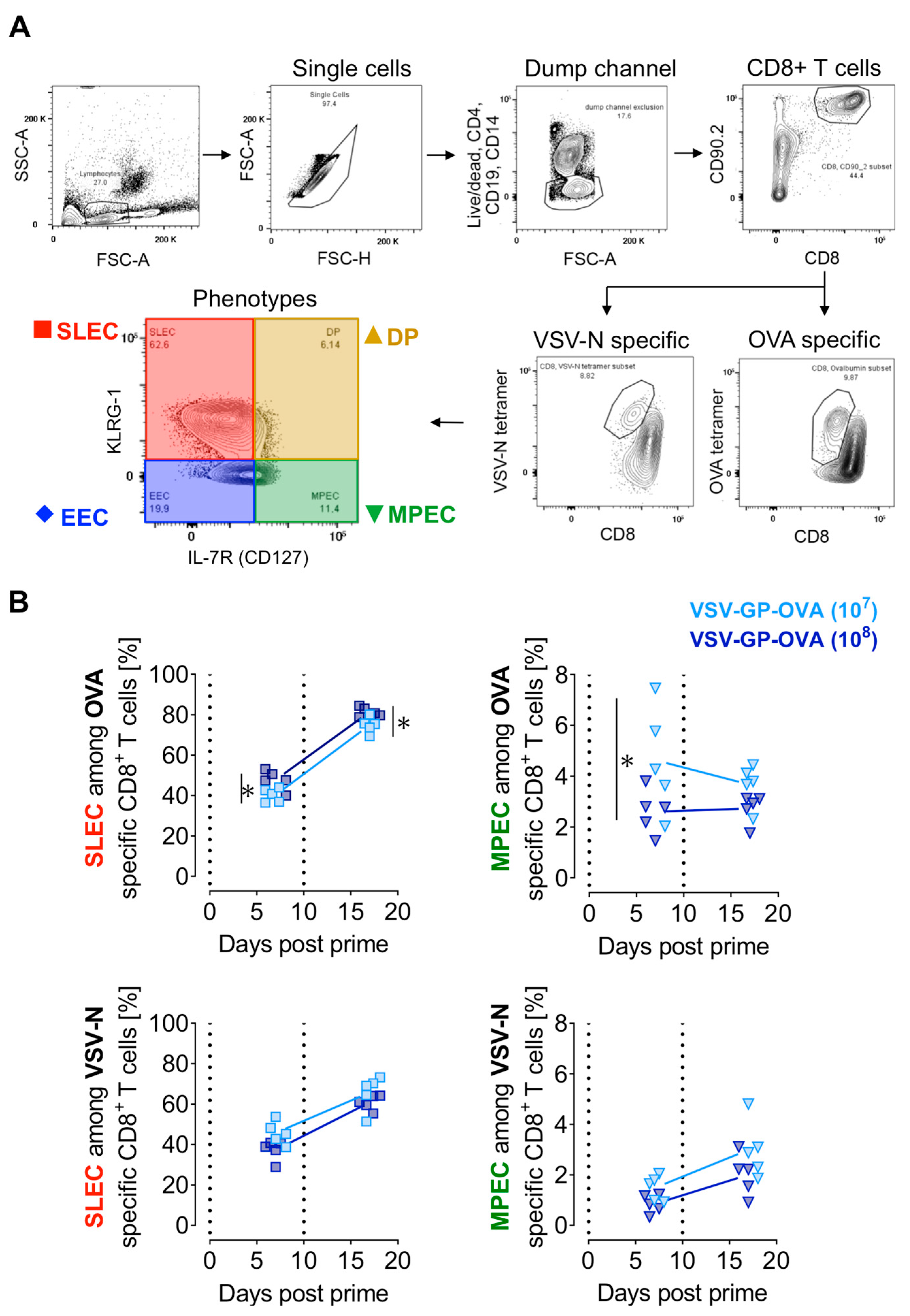
3.2. Prime-Boost Regimen and the Vaccine Dose Influence CD8+ T Cell Effector Subtypes
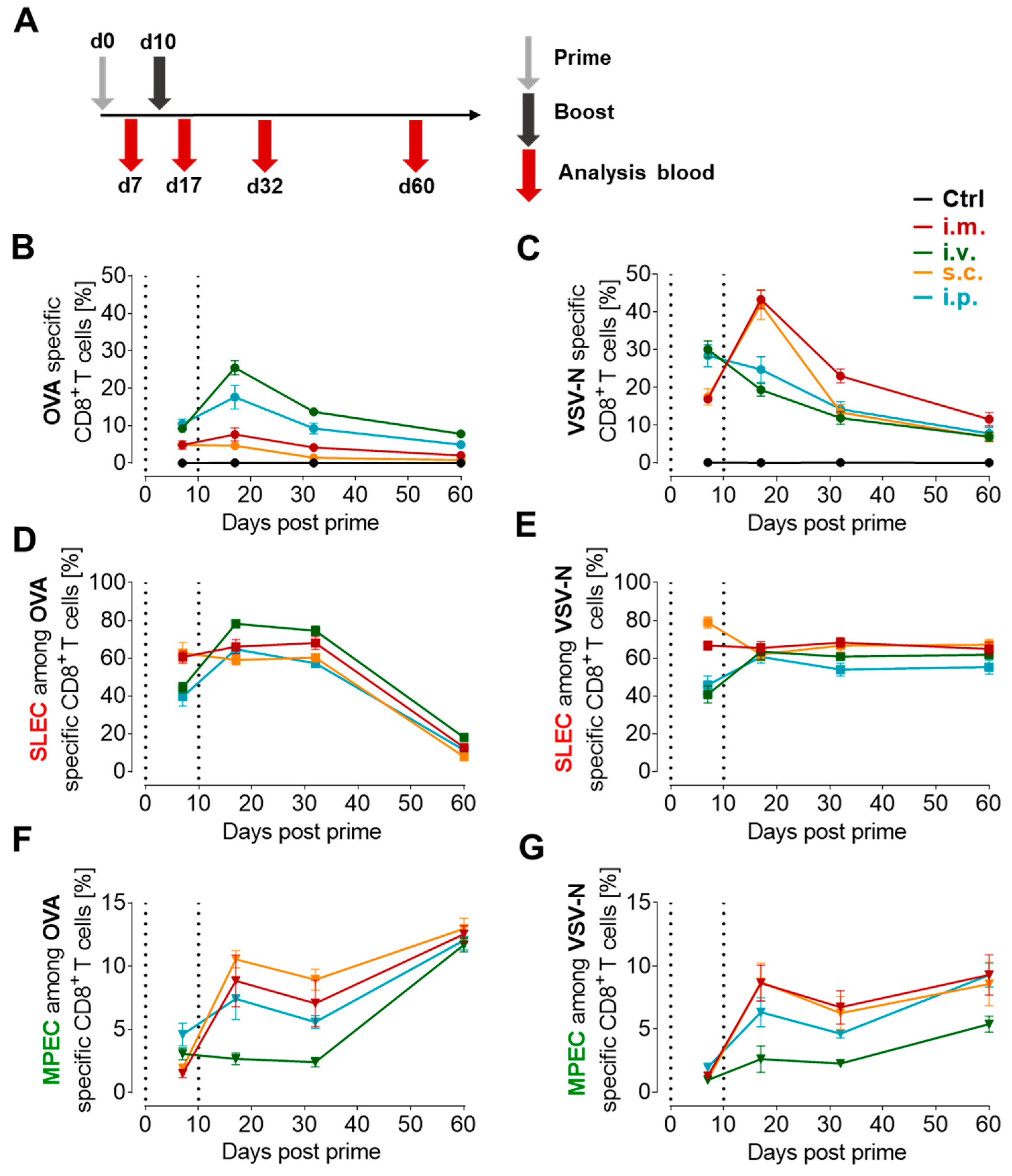
3.3. Proportion of Antigen-Specific CD8+ T Cells Depends on the Route of Immunization
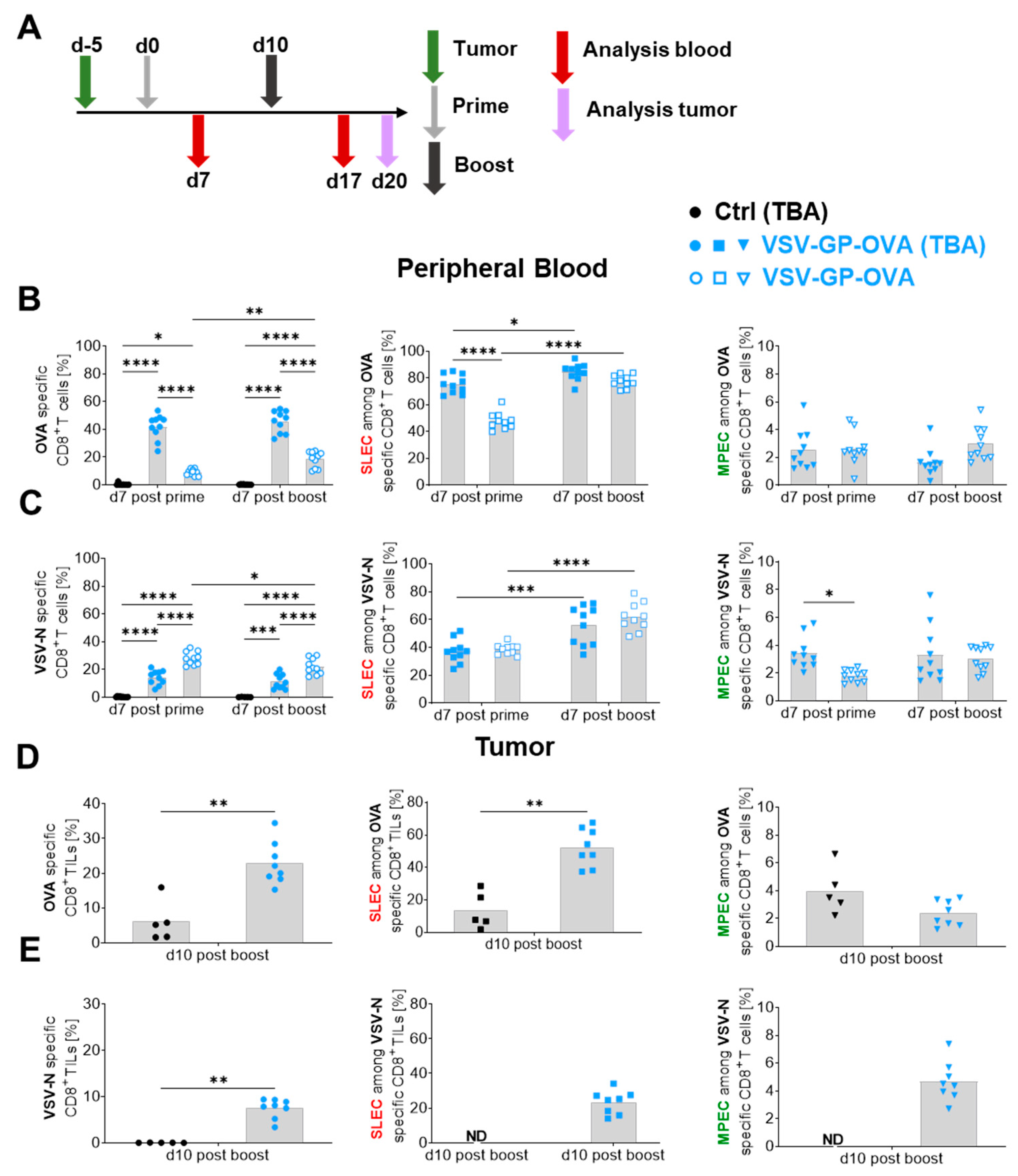
3.4. The Presence of a Target Antigen-Expressing Tumor Impacts Tumor-Antigen-Specific CD8+ T Cell Magnitudes as Well as Their Phenotype
3.5. Heterologous Vaccine Combination Increases Antigen-Specific CD8+ T Cell Levels and Changes the Effector Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, T.; Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E. Therapeutic cancer vaccine: Building the future from lessons of the past. Semin. Immunopathol. 2019, 41, 69–85. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef] [PubMed]
- Grayson, J.M.; Harrington, L.E.; Lanier, J.G.; Wherry, E.J.; Ahmed, R. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J. Immunol. 2002, 169, 3760–3770. [Google Scholar] [CrossRef]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. [Google Scholar] [CrossRef]
- Sellars, M.C.; Wu, C.J.; Fritsch, E.F. Cancer vaccines: Building a bridge over troubled waters. Cell 2022, 185, 2770–2788. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Carrette, F.; Surh, C.D. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin. Immunol. 2012, 24, 209–217. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1–more than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Plumlee, C.R.; Obar, J.J.; Colpitts, S.L.; Jellison, E.R.; Haining, W.N.; Lefrancois, L.; Khanna, K.M. Early Effector CD8 T Cells Display Plasticity in Populating the Short-Lived Effector and Memory-Precursor Pools Following Bacterial or Viral Infection. Sci. Rep. 2015, 5, 12264. [Google Scholar] [CrossRef]
- Herndler-Brandstetter, D.; Ishigame, H.; Shinnakasu, R.; Plajer, V.; Stecher, C.; Zhao, J.; Lietzenmayer, M.; Kroehling, L.; Takumi, A.; Kometani, K.; et al. KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 2018, 48, 716–729.e718. [Google Scholar] [CrossRef]
- Baharom, F.; Ramirez-Valdez, R.A.; Tobin, K.K.S.; Yamane, H.; Dutertre, C.A.; Khalilnezhad, A.; Reynoso, G.V.; Coble, V.L.; Lynn, G.M.; Mulè, M.P.; et al. Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol. 2021, 22, 41–52. [Google Scholar] [CrossRef]
- Karyampudi, L.; Lamichhane, P.; Scheid, A.D.; Kalli, K.R.; Shreeder, B.; Krempski, J.W.; Behrens, M.D.; Knutson, K.L. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 2014, 74, 2974–2985. [Google Scholar] [CrossRef]
- Obar, J.J.; Jellison, E.R.; Sheridan, B.S.; Blair, D.A.; Pham, Q.M.; Zickovich, J.M.; Lefrançois, L. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J. Immunol. 2011, 187, 4967–4978. [Google Scholar] [CrossRef]
- van Duikeren, S.; Fransen, M.F.; Redeker, A.; Wieles, B.; Platenburg, G.; Krebber, W.J.; Ossendorp, F.; Melief, C.J.; Arens, R. Vaccine-induced effector-memory CD8+ T cell responses predict therapeutic efficacy against tumors. J. Immunol. 2012, 189, 3397–3403. [Google Scholar] [CrossRef]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geiβ, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef]
- Porosnicu, M.; Quinson, A.M.; Crossley, K.; Luecke, S.; Lauer, U.M. Phase I study of VSV-GP (BI 1831169) as monotherapy or combined with ezabenlimab in advanced and refractory solid tumors. Future Oncol. 2022, 18, 2627–2638. [Google Scholar] [CrossRef]
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cao, W.; Salawudeen, A.; Zhu, W.; Emeterio, K.; Safronetz, D.; Banadyga, L. Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector. Pathogens 2021, 10, 1092. [Google Scholar] [CrossRef] [PubMed]
- Bresk, C.A.; Hofer, T.; Wilmschen, S.; Krismer, M.; Beierfuß, A.; Effantin, G.; Weissenhorn, W.; Hogan, M.J.; Jordan, A.P.O.; Gelman, R.S.; et al. Induction of Tier 1 HIV Neutralizing Antibodies by Envelope Trimers Incorporated into a Replication Competent Vesicular Stomatitis Virus Vector. Viruses 2019, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Wilmschen, S.; Schneider, S.; Peters, F.; Bayer, L.; Issmail, L.; Bánki, Z.; Grunwald, T.; von Laer, D.; Kimpel, J. RSV Vaccine Based on Rhabdoviral Vector Protects after Single Immunization. Vaccines 2019, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Belnoue, E.; Rossi, M.; Hofer, T.; Danklmaier, S.; Nolden, T.; Schreiber, L.M.; Angerer, K.; Kimpel, J.; Hoegler, S.; et al. A modular self-adjuvanting cancer vaccine combined with an oncolytic vaccine induces potent antitumor immunity. Nat. Commun. 2021, 12, 5195. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Rossi, M.; Carboni, S.; Di Berardino Besson, W.; von Laer, D.; Wollmann, G.; Derouazi, M.; Santiago-Raber, M.L. Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect. Cancers 2021, 13, 6107. [Google Scholar] [CrossRef] [PubMed]
- Tober, R.; Banki, Z.; Egerer, L.; Muik, A.; Behmüller, S.; Kreppel, F.; Greczmiel, U.; Oxenius, A.; von Laer, D.; Kimpel, J. VSV-GP: A potent viral vaccine vector that boosts the immune response upon repeated applications. J. Virol. 2014, 88, 4897–4907. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; den Boer, A.T.; Wagena, E.; Broen, K.; Dolstra, H.; de Boer, R.J.; Figdor, C.G.; Textor, J.; Friedl, P. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat. Commun. 2021, 12, 5217. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, L.M.; Urbiola, C.; Das, K.; Spiesschaert, B.; Kimpel, J.; Heinemann, F.; Stierstorfer, B.; Müller, P.; Petersson, M.; Erlmann, P.; et al. The lytic activity of VSV-GP treatment dominates the therapeutic effects in a syngeneic model of lung cancer. Br. J. Cancer 2019, 121, 647–658. [Google Scholar] [CrossRef]
- Cobleigh, M.A.; Bradfield, C.; Liu, Y.; Mehta, A.; Robek, M.D. The immune response to a vesicular stomatitis virus vaccine vector is independent of particulate antigen secretion and protein turnover rate. J. Virol. 2012, 86, 4253–4261. [Google Scholar] [CrossRef]
- Leveille, S.; Goulet, M.L.; Lichty, B.D.; Hiscott, J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J. Virol. 2011, 85, 12160–12169. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, J.; Urbiola, C.; Koske, I.; Tober, R.; Banki, Z.; Wollmann, G.; von Laer, D. The Oncolytic Virus VSV-GP Is Effective against Malignant Melanoma. Viruses 2018, 10, 108. [Google Scholar] [CrossRef]
- van Duikeren, S.; Arens, R. Predicting the efficacy of cancer vaccines by evaluating T-cell responses. Oncoimmunology 2013, 2, e22616. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, C.; Kasonta, R.; Lunemann, S.; Krähling, V.; Zinser, M.E.; Biedenkopf, N.; Fehling, S.K.; Ly, M.L.; Rechtien, A.; Stubbe, H.C.; et al. Dose-dependent T-cell Dynamics and Cytokine Cascade Following rVSV-ZEBOV Immunization. eBioMedicine 2017, 19, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Carretero-Iglesia, L.; Couturaud, B.; Baumgaertner, P.; Schmidt, J.; Maby-El Hajjami, H.; Speiser, D.E.; Hebeisen, M.; Rufer, N. High Peptide Dose Vaccination Promotes the Early Selection of Tumor Antigen-Specific CD8 T-Cells of Enhanced Functional Competence. Front. Immunol. 2020, 10, 3016. [Google Scholar] [CrossRef]
- Ols, S.; Yang, L.; Thompson, E.A.; Pushparaj, P.; Tran, K.; Liang, F.; Lin, A.; Eriksson, B.; Karlsson Hedestam, G.B.; Wyatt, R.T.; et al. Route of Vaccine Administration Alters Antigen Trafficking but Not Innate or Adaptive Immunity. Cell Rep. 2020, 30, 3964–3971.e3967. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Kumai, T.; Nagato, T.; Wu, J.; Salazar, A.M.; Celis, E. The route of administration dictates the immunogenicity of peptide-based cancer vaccines in mice. Cancer Immunol. Immunother. CII 2019, 68, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Tsao, J.; Schein, S.; Green, T.J.; Luo, M.; Zhou, Z.H. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 2010, 327, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Honke, N.; Shaabani, N.; Cadeddu, G.; Sorg, U.R.; Zhang, D.E.; Trilling, M.; Klingel, K.; Sauter, M.; Kandolf, R.; Gailus, N.; et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 2011, 13, 51–57. [Google Scholar] [CrossRef]
- Junt, T.; Moseman, E.A.; Iannacone, M.; Massberg, S.; Lang, P.A.; Boes, M.; Fink, K.; Henrickson, S.E.; Shayakhmetov, D.M.; Di Paolo, N.C.; et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 2007, 450, 110–114. [Google Scholar] [CrossRef]
- Backer, R.; Schwandt, T.; Greuter, M.; Oosting, M.; Jüngerkes, F.; Tüting, T.; Boon, L.; O’Toole, T.; Kraal, G.; Limmer, A.; et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 216–221. [Google Scholar] [CrossRef]
- Habbeddine, M.; Verthuy, C.; Rastoin, O.; Chasson, L.; Bebien, M.; Bajenoff, M.; Adriouch, S.; den Haan, J.M.M.; Penninger, J.M.; Lawrence, T. Receptor Activator of NF-κB Orchestrates Activation of Antiviral Memory CD8 T Cells in the Spleen Marginal Zone. Cell Rep. 2017, 21, 2515–2527. [Google Scholar] [CrossRef]
- Dambra, R.; Matter, A.; Graca, K.; Akhand, S.S.; Mehta, S.; Bell-Cohn, A.; Swenson, J.M.; Abid, S.; Xin, D.; Lewis, C.; et al. Nonclinical pharmacokinetics and biodistribution of VSV-GP using methods to decouple input drug disposition and viral replication. Mol. Therapy. Methods Clin. Dev. 2023, 28, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Al Shoyaib, A.; Archie, S.R.; Karamyan, V.T. Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies? Pharm. Res. 2019, 37, 12. [Google Scholar] [CrossRef]
- Shane, H.L.; Reagin, K.L.; Klonowski, K.D. The Respiratory Environment Diverts the Development of Antiviral Memory CD8 T Cells. J. Immunol. 2018, 200, 3752–3761. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Puorro, K.A.; Porgador, A.; Eisenlohr, L.C. The induction of virus-specific CTL as a function of increasing epitope expression: Responses rise steadily until excessively high levels of epitope are attained. J. Immunol. 1999, 163, 3735–3745. [Google Scholar] [CrossRef]
- Abraham, G.; Banerjee, A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1976, 73, 1504–1508. [Google Scholar] [CrossRef]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 150–158. [Google Scholar] [CrossRef]
- Kedl, R.M.; Kappler, J.W.; Marrack, P. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 2003, 15, 120–127. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Haring, J.S.; Harty, J.T. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8+ T cell response to infection. Immunity 2007, 26, 827–841. [Google Scholar] [CrossRef]
- Palmowski, M.J.; Choi, E.M.; Hermans, I.F.; Gilbert, S.C.; Chen, J.L.; Gileadi, U.; Salio, M.; Van Pel, A.; Man, S.; Bonin, E.; et al. Competition between CTL narrows the immune response induced by prime-boost vaccination protocols. J. Immunol. 2002, 168, 4391–4398. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.L.; Satpathy, A.T.; Wells, D.K. Bystander T cells in cancer immunology and therapy. Nat. Cancer 2022, 3, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Remmerswaal, E.B.M.; Hombrink, P.; Nota, B.; Pircher, H.; Ten Berge, I.J.M.; van Lier, R.A.W.; van Aalderen, M.C. Expression of IL-7Rα and KLRG1 defines functionally distinct CD8+ T-cell populations in humans. Eur. J. Immunol. 2019, 49, 694–708. [Google Scholar] [CrossRef]
- Martens, A.W.J.; Kavazović, I.; Krapić, M.; Pack, S.M.; Arens, R.; Jongejan, A.; Moerland, P.D.; Eldering, E.; van der Windt, G.J.W.; Wensveen, F.M.; et al. Chronic lymphocytic leukemia presence impairs antigen-specific CD8+ T-cell responses through epigenetic reprogramming towards short-lived effectors. Leukemia 2023, 37, 606–616. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofer, T.; Pipperger, L.; Danklmaier, S.; Das, K.; Wollmann, G. Characterization of the Anti-Viral and Vaccine-Specific CD8+ T Cell Composition upon Treatment with the Cancer Vaccine VSV-GP. Vaccines 2024, 12, 867. https://doi.org/10.3390/vaccines12080867
Hofer T, Pipperger L, Danklmaier S, Das K, Wollmann G. Characterization of the Anti-Viral and Vaccine-Specific CD8+ T Cell Composition upon Treatment with the Cancer Vaccine VSV-GP. Vaccines. 2024; 12(8):867. https://doi.org/10.3390/vaccines12080867
Chicago/Turabian StyleHofer, Tamara, Lisa Pipperger, Sarah Danklmaier, Krishna Das, and Guido Wollmann. 2024. "Characterization of the Anti-Viral and Vaccine-Specific CD8+ T Cell Composition upon Treatment with the Cancer Vaccine VSV-GP" Vaccines 12, no. 8: 867. https://doi.org/10.3390/vaccines12080867