Abstract
Chagas disease (CD) treatment and vaccine development are critical due to the significant health burden caused by the disease, especially in Latin America. Current treatments include benznidazole and nifurtimox, which are most effective in the acute phase of the disease but less so in the chronic phase, often with significant side effects. Here, using the available literature, we summarize the progress in vaccine development and new treatments that promise to reduce CD incidence and improve the quality of life for those at risk, particularly in endemic regions. New treatment options, such as posaconazole and fexinidazole, are being explored to improve efficacy and reduce adverse effects. Vaccine development for CD remains a high priority. The complex life stages and genetic diversity of Trypanosoma cruzi present challenges, but several promising vaccine candidates are under investigation. These efforts focus on stimulating a protective immune response through various innovative approaches.
1. Introduction
Chagas disease (CD), also known as American Trypanosomiasis, is caused by the protozoan parasite Trypanosoma cruzi. CD has become a major public and social health problem in Latin America and is considered a neglected tropical disease by the World Health Organization [1]. CD affects approximately 8–10 million people worldwide, with more than 100 million people exposed to the risk of infection [2]. However, these estimates could be higher due to underreporting and limited access to healthcare in some regions.
Chagas disease was initially considered endemic to certain regions of Latin America, such as Bolivia, Argentina, Paraguay, Equator, El Salvador, and Guatemala, with the highest rate of infected individuals occurring in these regions [2,3]. However, due to increased globalization and the resulting higher migratory movements, CD has become a global health concern, including non-endemic countries such as the United States, Spain, Japan, and Australia. This is mainly due to the lack of monitoring and thorough screening of blood banks and the inexperience of health professionals in the diagnosis and management of CD, which contribute to non-vectorial routes of infection such as organ transplant and congenital transmission [4,5].
The life cycle of T. cruzi involves several stages and two main hosts: a triatomine insect (commonly known as the “kissing bug”) and a mammalian host, such as humans or other mammals [6]. The cycle begins when an infected triatomine bug takes a blood meal from a mammalian host, ingesting trypomastigotes present in the host’s blood. In the bug’s midgut, the trypomastigotes transform into epimastigotes, which multiply through binary fission. The epimastigotes then migrate to the hindgut, where they transform into metacyclic trypomastigotes, the infective form for mammals. Subsequently, the triatomine bug defecates during or after taking a blood meal, releasing metacyclic trypomastigotes in its feces. The parasites enter the mammalian host through mucous membranes, broken skin, or the bite wound itself. Once inside the host, the metacyclic trypomastigotes invade various cells (commonly muscle cells, macrophages, and nerve cells) and transform into intracellular amastigotes. The amastigotes multiply by binary fission within the host cells, eventually transforming back into trypomastigotes. The host cells burst, releasing trypomastigotes into the bloodstream, where they can infect new cells or be ingested by another triatomine bug, continuing the cycle [7,8].
The disease has two clinical phases: an acute phase and a chronic phase. The acute phase usually lasts 4–8 weeks and is characterized by the presence of amastigotes in cells adjacent to the subcutaneous tissue and circulating cells such as leukocytes and macrophages, allowing the parasite to spread through the lymphatic and blood system, lodging in other tissues [4]. This phase of the disease is marked by detectable parasitemia and inflammation caused by tissue parasitism [9]. It is usually asymptomatic or has non-specific symptoms that may include fever or flu-like symptoms that can disappear without treatment, compromising the diagnosis of the disease [4]. Patients not effectively treated during the acute phase of the disease may progress to the chronic phase after 2–3 months of infection, when the parasitemia is usually undetectable by the conventional diagnostic methods that are available [9]. Ten to thirty years after the acute phase, 60–70% of patients develop the indeterminate form of the disease and may not show any clinical signs of CD throughout their lifespan. On the other hand, 30–40% of patients evolve to specific chronic conditions of CD, with clinical manifestations of the disease’s cardiac, digestive, or cardiodigestive forms [10].
In Latin America, CD poses a significant economic burden regarding healthcare costs and the socioeconomic impact on affected individuals and communities [11,12]. It is a multifactorial load that includes increased health care costs due to long-term management and treatment, productivity loss since it affects primarily working-age individuals who may become disabled or die prematurely, and other factors such as the social and economic impact on the affected individuals, contributing to economic inequality perpetuation. Currently, CD is responsible for the loss of approximately 750,000 workdays due to premature deaths and USD 1.2 billion in lost productivity each year [12,13]. Globally, this disease is estimated to result in 50,000 deaths annually and between 1.18–5.85 Disability-Adjusted Life Years (DALYs) [14], implying a global economic burden of around USD 7 billion per year due to both the cost of treatment and financial losses caused by lost productivity by or morbidity of infected people [11].
Therefore, the importance of developing treatments and/or vaccines for CD cannot be overstated. Effective treatments can reduce long-term management needs and prevent disability or premature death among working-age individuals, thereby improving their quality of life and reducing the global burden of the disease, which currently affects millions of people worldwide. Prophylactic or therapeutic vaccines would provide a preventative measure, potentially eradicating or significantly reducing the incidence of CD in endemic and newly endemic areas. Thus, in this review, we focus on current and prospective new treatments and the perspective for either prophylactic or therapeutic vaccine candidates for CD.
2. Treatment
2.1. Current Treatment
The etiological treatment of CD mainly aims to eliminate the T. cruzi parasite, consequently improving patients’ clinical outcomes and interrupting the disease transmission cycle [15]. Only two drugs are available to treat the infection: Benzidazole (BZN) and Nifurtimox (NFX), which have been consistently used to treat the disease over the last 60 years [3]. BZN is currently on the WHO list of essential medications [1,16] and is part of a group of nitro heterocyclic compounds containing one or more nitro groups linked to an aromatic ring [15]. This substance is a prodrug which exerts its effect after the type I trypanosomal nitro-reductase enzyme activation present in T. cruzi and other protozoa, producing reactive metabolites that have a trypanocidal effect on the intra- and extra-cellular forms of the parasite [15,17]. On the other hand, NFX is a nitrofuran compound whose mechanism of action consists of producing reduced oxygen metabolites—nitroanions—after drug activation by the parasite’s nitroreductases in the presence of oxygen. This leads to the production of free radicals that block DNA synthesis and accelerate DNA and RNA degradation, vital cellular components for T. cruzi [12,17,18]. However, their efficacy and safety data are not ideal, as both drugs have a high rate of adverse effects [17].
Treatment is effective during the acute phase of the disease—showing a 60–85% cure rate—in addition to reducing the severity of symptoms, shortening the clinical course, and decreasing the detection of parasitemia. Unfortunately, infected people are not always correctly diagnosed or treated because their broad symptoms are often mistaken for symptoms of other more common diseases [17]. Recent data has shown that less than 1% of infected patients are diagnosed and treated correctly [12], reaffirming the need for an accurate initial diagnosis so that therapeutic interventions are carried out early [3]. Conversely, in 1983 a panel of experts recommended not treating patients in the chronic phase. This recommendation was based on limited evidence of the effectiveness of etiological treatment in this phase and the belief that the characteristic symptoms of chronic CD resulted from an exacerbated immune response unrelated to the parasite. However, there was irrefutable evidence that parasite persistence acted as a triggering factor for chronic CD pathology [15]. Since then, the efficiency of treatment in the chronic phase has been under debate, as studies have shown that treatment in the chronic phase could be associated with delayed progression of the disease, although only 15% of patients show negative seroconversion after ten years [19]. Therefore, several clinical trials have been carried out with both drugs (Table 1) to establish whether the pharmacotherapy traditionally used for treating CD is effective in negative serological and molecular tests or capable of retarding the progression of the chronic disease [17].

Table 1.
Clinical trials for Chagas disease using Benznidazole or Nifurtimox.
2.2. New Treatments
Although there was significant improvement in new drug discovery regarding other trypanosomatid diseases, such as African trypanosomiasis and leishmaniasis, developing new medications for CD has proven more challenging [28]. Evaluation of new treatments for CD focuses on reducing the side effects of the current treatment (BZN and Nifurtimox), reducing parasite burden, and curing or preventing the chronic phase of CD, but there have been significant challenges in finding molecules that can achieve complete parasite clearance.
Recently, Padilla et al. identified a highly effective compound of a class of benzoxaboroles, AN15368, for treating CD. The results showed a uniform infection cure in non-human primates with long-term naturally acquired infections of diverse T. cruzi genetic types [29]. Results indicate that AN15368 is safe, elicits no significant side effects, and is more effective than existing drugs, with clinical trials expected to start in the next few years. Since 2009, the Drugs for Neglected Diseases initiative (DNDi) has been raising efforts for massive drug screening in order to find new drug candidates for CD. However, only the benzoxarobole compound DNDI-6148 showed promising results and has progressed to phase I studies [30].
Fexinidazole was approved in 2018 for treating sleeping sickness, and it is a nitroimidazole compound that has shown promise as a potential treatment for CD as an oral treatment of acute and chronic experimental CD [31]. Ravuconazole (E1224) is an antifungal drug that has demonstrated activity against T. cruzi in an experimental acute murine model. In the early stages of infection, administering optimal doses of E1224-benznidazole combination treatment led to a 100% cure rate. However, this approach was unable to eradicate an already well-established disease. Nonetheless, the combination therapy showed significant benefits, as it further reduced parasitemia compared to using either drug alone [32]. New drug formulations have also been tested for CD (Table 2), mainly in the chronic stage of infection.
Table 2.
Clinical trials for Chagas disease proposing the use of new medications.
3. Vaccines
As CD is a significant public health concern in Latin America, developing a preventive/therapeutic vaccine would have an enormous economic impact in the region [14]. An effective vaccine could provide long-term protection against the T. cruzi parasite, preventing new infections and reducing the costs associated with diagnosis, treatment, and management of the disease, consequently reducing morbidity/mortality and the burden on public health costs [38]. For example, up to USD 1.2 million would be saved if a therapeutic vaccination strategy was applied in pregnant women to avoid the progression of CD in infected children [39].
However, the development of a vaccine for CD faces several challenges that need to be addressed:
- Complex parasite life cycle: T. cruzi, has a complex life cycle involving different stages and multiple forms of the parasite, and each stage may require a different type of immune response for effective control and eradication [6].
- Limited understanding of protective immunity: The precise immune response required to confer protection against CD is not fully understood yet [40,41]. Identifying the key immune mechanisms involved in controlling the infection and developing vaccines that elicit those specific responses is essential to creating an effective CD vaccine.
- Lack of predictive animal models: Animal models used in preclinical studies may not fully recapitulate the complex dynamics of CD in humans. Therefore, it can be challenging to accurately predict the effectiveness of vaccine candidates in humans based on animal studies alone [42,43].
- Lack of surrogate markers of protection: To date, no established surrogate markers can reliably predict vaccine efficacy against CD [44]. The absence of such markers makes it challenging to assess the efficacy of vaccine candidates in clinical trials and may require long-term follow-up to determine their effectiveness.
- Limited commercial interest: CD primarily affects marginalized and economically disadvantaged populations, predominantly in Latin America. The lack of financial incentives for pharmaceutical companies has historically hindered the development of vaccines for neglected tropical diseases. Public–private partnerships and alternative funding mechanisms might be necessary to overcome this challenge [45].
- Regulatory and manufacturing challenges: Vaccine development involves navigating complex regulatory processes and scaling production to meet global demand [46]. Regulatory approvals, manufacturing infrastructure, and cost-effectiveness are significant challenges that must be addressed to ensure access to an affordable and widely available CD vaccine [45].
Nevertheless, vaccines are one of the most effective tools for preventing infectious diseases [47]. Several preclinical vaccine candidates for CD have been investigated in animal models. These candidates aim to elicit an immune response against T. cruzi, with humoral and cell-mediated immune responses to extracellular trypomastigotes and intracellular amastigotes (Figure 1).
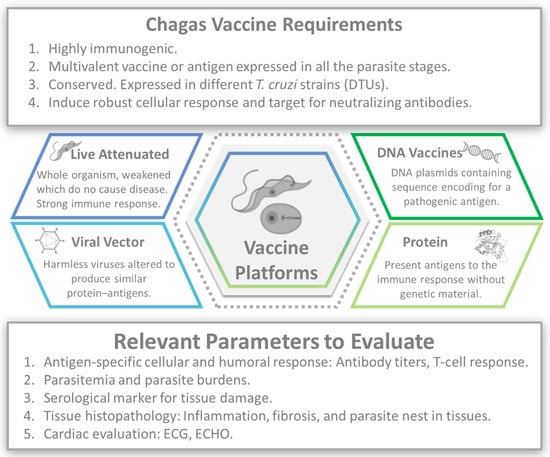
Figure 1.
Chagas vaccine development. Image created with the assistance of BioRender.
A wide array of vaccination strategies has been conducted to date, including, but not limited to, nucleic acids, protein subunits, attenuated vaccines, and nanoparticles, which have been assessed in mice with promising results [48,49].
3.1. Attenuated Vaccines
Attenuated vaccines have been instrumental in controlling and eliminating several infectious diseases, including polio, yellow fever, varicella, rotavirus, influenza, measles, mumps, and rubella. These types of vaccines induce strong and lasting immunity. Despite their effectiveness, they pose challenges, especially in immunocompromised individuals. However, these vaccines are highly effective in veterinary medicine, contributing significantly to the control and prevention of animal diseases, which make a great type of vaccines for controlling CD. Live attenuated vaccines involve modifying the parasite to reduce its virulence while maintaining its immunogenicity, which has been successfully tested in other protozoa, including Trypanosoma brucei, Leishmania major, and Plasmodium.
In the case of CD, the genetic manipulation of T. cruzi has been more challenging and only few attenuated-parasite approaches have been used as vaccines in preclinical models. Some of the vaccine candidates include an attenuated parasite with the deletion of the lyt-1 gene and the parasite factor involved [50]. A second approach was the deletion of the surface glycoprotein 72 (gp72), which affects the morphology of the parasite in multiple stages of the life cycle [51]. A third approach deletes the enoyl-coenzyme A (CoA) hydratase 1 and 2 genes (ECH1+/− ECH2−/−), two enzymes involved in the amastigote metabolism. The last approach is the deletion of the bifunctional enzyme dihydrofolate reductase-thymidylate synthase (DHFR-TS) [52]. These attenuated parasites fail to establish persistent infection, can induce strong immune responses, and protect against subsequent T. cruzi challenges through an increment in the CD8+ T-cell immune response [53]. One of the biggest challenges of attenuated vaccines, particularly in the case of attenuated parasites, is obtaining sufficient quantities of the parasite in consistent quality for use in vaccinations. Variability in parasite quality can affect the immune response elicited by the vaccine, leading to inconsistent protection. However, the use of attenuated vaccines in animals is often more acceptable and practical than in humans due to a combination of risk tolerance, economic considerations, and regulatory environments. All of these candidates could potentially decrease the number of reservoirs that infect triatomine bugs.
3.2. DNA Vaccines
DNA vaccines work by introducing a small piece of the pathogen’s DNA into the body, which encodes specific proteins or antigens. These vaccines have been widely explored as a potential approach for developing a vaccine against CD, as the produced antigens stimulate an immune response, triggering the production of antibodies and activating immune cells to recognize and neutralize the pathogen. To date, multitudes of T. cruzi DNA vaccine studies have been conducted (Table 3) using several well-characterized T. cruzi virulence factors. For example, cruzipain is a significant cysteine-protease enzyme and GP82 is a surface glycoprotein of T. cruzi that plays a role in host cell invasion [54]. The amastigote surface protein-2 (ASP-2) and TcG1 are T. cruzi antigens expressed in the parasite’s metacyclic trypomastigote form, which is involved in the parasite’s entry into host cells [55]. When mice were immunized with recombinant Trypanosoma cruzi small surface protein 4 (rTcSSP4), they became more susceptible to trypomastigote infection, and this resulted in high mortality rates. On the other hand, when mice were immunized with a eukaryotic expression plasmid containing the TcSSP4 cDNA, the acute phase of infection was effectively controlled. The difference in outcomes between the two forms of TcSSP4 immunization highlights the importance of understanding the type of immune response generated by a vaccine candidate and how it influences protection against the target pathogen [56]. Finally, other studies have been used trans-sialidases, with the administration of CpG or the cytokine IL-15, effectively in mice to reduce mortality and chronic disease [57,58,59].
Table 3.
An overview of the DNA vaccines and their efficacy for prophylaxis of Chagas disease in mouse models. Only the studies that assessed most of the parameters listed below were selected and referenced in the table.
3.3. Viral Vector
Viral vectors are a type of vaccine that uses a harmless virus to deliver genetic material into the human body’s cells (Table 4). The genetic material typically encodes a specific antigen from T. cruzi to stimulate an immune response without causing the disease. These viral vectors have been used as gene delivery systems for recombinant vaccines and gene therapy applications, since unlike subunit vaccines they can elicit a humoral response without needing an adjuvant. Other advantages of the viral-vector vaccines are the strong and durable immune responses, versatility in design, genetic stability, and production efficiency. However, viral-vector vaccines have several challenges and potential problems, including pre-existing immunity that can neutralize the viral vector vaccine and reduce efficacy and safety concerns, including insertional and mutagenic concerns. Virus vectors expressing the T. cruzi antigens TSSA, TS, and ASP-2 induce specific antibody-secreting B cells and cytotoxic T cells with elevated levels of IFN-γ, resulting in reduced parasitemia and an increment in survival [60,61].
Table 4.
An overview of the viral vector vaccines and their efficacy for prophylaxis of Chagas disease in mouse models. Only the studies that assessed most of the parameters listed below were selected and referenced in the table.
3.4. Recombinant Protein or Peptide Vaccines
Recombinant protein vaccines use purified or synthesized parasite proteins to induce an immune response, and similar to the T. cruzi DNA vaccines, protein vaccines have been investigated as potential candidates for generating a vaccine against CD. These include well-characterized T. cruzi antigens capable of priming the immune system with potent humoral and cellular immunity to confer protection [48,62]. Some of these T. cruzi antigens include trans-sialidase, amastigote surface proteins, or cruzipain. In addition to whole-protein vaccines, subunit vaccines are also potential candidates and typically consist of specific protein fragments or peptides derived from the parasite. These vaccines aim to focus the immune response on crucial antigenic regions of the parasite while reducing potential side effects [63]. Some preclinical studies have evaluated subunit vaccines containing peptides or recombinant protein fragments derived from T. cruzi, which have shown promising results in terms of immunogenicity and protection (Table 5).
Table 5.
An overview of the recombinant protein vaccine platform and their efficacy against Chagas disease in mouse models. Only the studies that assessed most of the parameters listed below were selected and referenced in the table.
Promising results have been reported from the surface proteins, Tc24 and trypomastigote small surface sntigen-1 (TSA-1), using aluminum phosphate as an adjuvant, and were therapeutic (BALBc, c57BL/6 and ICR) and/or preventive (BALBc) in their treatment outcome. BALBc and c57BL/6 mice with acute infection, and ICR mice with chronic infection showed and induction of balanced humoral and Th1/Th2/Th17 cellular responses have a protective efficacy against T. cruzi [69,70].
3.5. Glycoconjugates
The glycocalyx of T. cruzi consists of abundant, complex, highly variable, and immunogenic glycosylphosphatidylinositol (GPI)-anchored glycoproteins and glycolipids [72]. These include mucins, mucin-associated surface proteins (MASPs), TS/gp85 glycoproteins, and glycoinositolphospholipids, which vary throughout the parasite’s life-cycle stages [73]. Among the glycoproteins present on trypomastigotes, the GPI-anchored mucins (tGPI-mucins) carry the immunodominant glycotope Galα1,3Galβ1,4GlcNAc (Galα3LN) and several branched α-Gal-terminating O-glycans that have not been fully characterized yet. The presence of these α-Gal glycotopes stimulates the production of high levels of CD-specific anti-α-Gal antibodies in CD patients. These antibodies play a protective role by effectively controlling the parasitemia during the disease’s acute and chronic phases [74,75] and are found in abundance among patients from endemic and non-endemic countries. These molecules aim to induce an immune response against the parasite to provide protection against the disease. These proteins have effectively protected against CD [76] and leishmaniasis [77] and could potentially serve as vaccine candidates for acute and chronic CD (Table 6).
Table 6.
An overview of the recombinant protein vaccine platform and its efficacy against Chagas disease in mouse models. Only the study that assessed most of the parameters listed below was selected and referenced in the table.
3.6. Multivalent Vaccines
A multivalent vaccine combines multiple antigens from the same or different pathogens into a single vaccine formulation. This approach is often used to target multiple strains or stages of a pathogen and enhance the vaccine’s overall effectiveness [78]. For example, TcG1–TcG8 are 8 vaccine antigens phylogenetically conserved in clinically important strains of T. cruzi that are expressed in the infective (trypomastigote) and intracellular (amastigote) parasite stages. These vaccine candidates elicited varying levels of lytic antibody response and Th1 cytokines (IFN-γ), a property associated with immune control of T. cruzi. TcG1-, TcG2-, and TcG4-encoded antigens were expressed on the plasma membrane of the mammalian stages of T. cruzi (trypomastigote/amastigote) and elicited significant levels of anti-parasite lytic antibody responses in a micNA-prime/protein-boost subunit vaccine (TcVac2) [79].
DNA vaccines composed of ASP-1, ASP-2, and TSA-1 have been shown to provide partial protection from lethal T. cruzi infection, and importantly, they can significantly reduce the severity of chronic CD. Glycosylphosphatidylinositol (GPI)-anchored proteins from T. cruzi—ASP-1, ASP-2, and TSA-1—that are the targets of CD8+ cytotoxic T lymphocytes (CTLs) and that induce strong antibody responses in infected mice and humans belong to the T. cruzi trans-sialidase family. Trans-sialidases are enzymes that transfer sialic acid residues from host glycoconjugates to the parasite’s own surface glycoproteins, allowing the parasite to modulate host immune responses and evade the immune system [80]. However, it’s also important to note that despite controlling T. cruzi infection and the beneficial effects on CD severity, the vaccines using the three genes tested in the study failed to inhibit infection or eliminate parasites from infected animals completely (Table 7). Moreover, they were unable to prevent death from infection in 100% of vaccinated animals. These results indicate that while the genetic vaccines targeting these specific proteins look promising and positively impact disease progression, they are not fully effective in providing complete protection against T. cruzi infection or eradicating the parasite in all cases [81].
Table 7.
An overview of the multivalent vaccines and their efficacy against Chagas disease in mouse models. Only the studies that assessed most of the parameters listed below were selected and referenced in the table.
3.7. Heterologous Vaccines
Heterologous vaccination refers to the administration of different types of vaccines for the primary and booster doses of a vaccination schedule. In contrast, homologous vaccination involves using the same vaccine for both the initial and booster shots. Despite potentially raising development costs and complicating production, heterologous vaccination holds the promise of providing superior immunity. As a result, it becomes particularly compelling for vaccine development efforts targetting complex pathogens like parasites [83]. A heterologous strategy can optimize T-cell responses to elicit robust polyfunctional T-cell responses, especially considering that multiple boosts in the same immunization site may lead to T-cell sequestration (Table 8). This approach can effectively trigger cell-mediated immunity against parasites [84].
Table 8.
An overview of the heterologous schemes and their efficacy in Chagas disease in mouse models. Only the studies that assessed most of the parameters listed below were selected and referenced in the table.
3.8. mRNA Vaccines
Developing a vaccine for CD poses several challenges due to the parasite’s life cycle complexity and the variety of immune responses required for protection. Nonetheless, researchers have made progress in exploring potential vaccine candidates and studying the immunological responses needed to combat the infection. Technological advancements in vaccine development provide immunity against infectious diseases by utilizing various cutting-edge technologies and methodologies, including mRNA vaccines [88]. The Tc24 antigen was recently evaluated in a heterologous mRNA prime/protein boost strategy, showing promising results that include an improvement in the humoral and cellular responses with robust polyfunctional cells, with the equivalent-balanced Th1/Th2/Th17 secreted-cytokine profile required for complete protection against T. cruzi [89]. However, efficacy data is needed to understand if the immune profile obtained is able to control the infection. Recently, a study explored the development and potential of mRNA-based vaccines targeting T. cruzi antigens Tc24 and ASP-2 as a therapeutic option for CD, showing promising results in reducing parasite burdens and cardiac inflammation in a chronic mouse model [88].
Several notable challenges hinder the widespread clinical application and large-scale mRNA vaccine immunization programs, particularly for the Latin American region. These obstacles include a lack of quality control measures, inconsistent purification standards, insufficient manufacturing capabilities, storage inadequacies, management supervision issues, and suboptimal biocompatibility [90,91].
4. Vaccine-Linked Chemotherapy
An interesting approach for treating CD is vaccine-linked chemotherapy. This approach combines the antiparasitic drug BZN to reduce parasite load with the vaccine antigen, boosting the cellular and humoral immune responses. The first studies focused on vaccine-linked chemotherapy evaluated the recombinant protein vaccine Tc24-C4 + TLR4 adjuvant and showed promising results not only in a reduction of parasite burden, but also in a reduction of cardiac inflammation and fibrosis. The scheme has been tested on acute and chronic infection models using the H1 strain of T. cruzi. This strategy allowed a reduction in the dose of BZN to 25 mg/kg per mouse, which will benefit the patients by minimizing the side effects of the treatment and making it more tolerable [92,93,94,95]. Testing of this strategy in a chronic model of infection showed improved cardiac structure and function and cardiac metabolism [94,95]. Similarly, the recombinant protein vaccine antigen TS + ISPA was evaluated in conjunction with the curative dose of BZN (100 mg per mouse) and showed a reduction in parasite burdens and reduced cardiac arrhythmias [96]. These studies showed that combining vaccine-linked chemotherapy has enhanced efficacy in BNZ treatment and offers a dose-sparing approach that may improve overall cardiac health and clinical outcomes.
5. Discussion
The development of new treatments and a vaccine for CD has faced numerous scientific and logistical challenges. Traditional treatments with benznidazole and nifurtimox are plagued by side effects and limited efficacy in chronic stages of the disease, underscoring the need for better therapeutic options [97]. As highlighted in this review, most clinical trials for CD focus on optimizing new regimens of the already extensively used medications, Benznidazole and Nifurtimox [97]. Benznidazole, a nitroimidazole derivative, has been the cornerstone of Chagas disease treatment for decades due to its efficacy in the acute and early chronic stages of the disease. Nifurtimox, another nitrofurane-based compound, similarly offers benefits in these stages [17]. However, both drugs are associated with significant side effects, such as gastrointestinal disturbances and neurological symptoms, which often lead to treatment discontinuation [26].
Recent trials have aimed to mitigate these adverse effects by experimenting with reduced dosages and shorter treatment durations. For instance, new regimens have included lower doses of Benznidazole over shorter periods, which have shown promise in maintaining efficacy while improving patient tolerance [20,21,25,26]. Additionally, combination therapies involving Benznidazole or Nifurtimox with other drugs are being explored to enhance therapeutic outcomes and reduce the parasite load more effectively [33,34,37]. The potential of nitroheterocyclic compounds like fexinidazole and its metabolites demonstrated superior efficacy in murine models compared to benznidazole [31,98]. Furthermore, CYP51 inhibitors such as posaconazole [36,99] and ravuconazole [33] have shown promising results in preclinical trials. The combination of nitroheterocyclic drugs with azoles was particularly effective, suggesting that multi-drug regimens could improve therapeutic outcomes. The application of nanomedicine, such as formulations in nanocarriers [100,101], also displayed outstanding efficacy against various T. cruzi strains, indicating a potential path forward for overcoming drug resistance and enhancing treatment efficacy that could potentially be applied to other molecules. Thus, these efforts are crucial for increasing the accessibility and effectiveness of Chagas disease treatment, particularly in resource-limited settings where the disease is most prevalent [102].
On the other hand, the development of both prophylactic and therapeutic vaccines has been a focal point of research to combat this disease, but despite significant efforts, an effective vaccine remains elusive due to the complex life cycle of T. cruzi and the varied immune responses required to control its different stages. Early attempts at inoculation of mice with attenuated strains showed only partial immunity. While many vaccination strategies have been explored, including the use of attenuated strains, cultural forms, and molecular components of T. cruzi, none have achieved sterile immunity [103]. Several studies have made significant strides in understanding the immunological mechanisms of T. cruzi. For instance, the importance of CD8+ T cells and IFN-γ in controlling T. cruzi infection and the discovery of key antigen candidates such as TS, ASP-2, Tc24, and the α-Gal epitope, which have shown promise in inducing protective immunity in murine models [103]. Additionally, the potential use of recombinant adenoviruses encoding these antigens to induce long-term protective immunity in animal models was also explored [48].
Despite these advances, translating findings from animal models to human applications remains a significant hurdle. Animal studies have limitations in predicting human responses due to biological differences between species, leading to vaccine efficacy and safety discrepancies. The predictive value of animal models is often questioned, with a growing consensus that alternative methods, such as specific in vitro models and organ-on-chip technologies, might offer better predictive capabilities [43,104].
6. Perspectives
Over the course of several decades, the relentless pursuit of a CD vaccine has persisted, yet we find ourselves still lacking an approved vaccine for broad-scale immunization programs, especially in regions where CD is endemic [105]. It is imperative to underscore that vaccine development is a complex process, and it may take time before an effective vaccine for CD becomes available. However, as highlighted in this review, developing a CD vaccine is a highly dynamic and vigorously pursued field of research, and several potential vaccine candidates are being continuously investigated [63].
Given the complexity of the T. cruzi parasite, which includes different life stages and a broad genetic diversity that might influence vaccination and treatment effectiveness, a multifaceted approach might be required to develop a successful CD vaccine. Additionally, efforts towards the advancement of vaccine development to preclinical and clinical trials are needed to move the best candidates up to the next level and closer to a regulatory agency-approved vaccine to be used in the patients effectively. This involves collaborative efforts of international organizations, governments, research institutions, and industry to accelerate this much-needed tool against CD [48].
The use of vaccines against T. cruzi in veterinary medicine could provide significant benefits, especially for domestic and wild animals that serve as reservoirs for the parasite. For example, rabies vaccines for domestic animals have been crucial in reducing transmission to humans through animal bites, with widespread implementation decades before human vaccines were developed [106]. This underscores the proactive role of veterinary vaccines in mitigating zoonotic disease risks and influencing subsequent human vaccine developments. Vaccinating animals can help reduce the transmission of T. cruzi to humans and decrease the prevalence of Chagas disease in endemic areas. By controlling the disease in animal populations, the risk of human infection can be significantly diminished, particularly in rural and peri-urban areas where human–animal interactions are frequent.
Veterinary vaccination programs can be integrated with other control measures, such as vector control and improved housing conditions, to create a comprehensive strategy against Chagas disease. Veterinary vaccines against T. cruzi will likely be implemented more quickly than human vaccines, and this rapid deployment can play a crucial role in controlling the disease in animal populations, thereby reducing the transmission risk to humans. While veterinary vaccines alone may not eliminate Chagas disease, they are a critical component of an integrated approach to managing and potentially eradicating this public health threat.
Furthermore, we must emphasize the necessity of unwavering commitment to the advancement of vaccine development. Transitioning promising research findings and potential candidates from the laboratory bench to the real-world context of preclinical and clinical trials is a pivotal step in the journey to provide a safe and effective CD vaccine for patients, especially those at risk in endemic areas. This dedicated and rigorous approach ensures that we navigate the challenging terrain of vaccine development with due diligence, bringing us closer to a solution for developing a successful CD vaccine.
Author Contributions
Conceptualization, P.S.G.F., K.M.J. and C.P.; writing—original draft preparation, P.S.G.F. and C.P.; writing—review and editing, P.S.G.F., K.M.J. and C.P. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the support of Texas Children’s Hospital, which supports the development of a vaccine against Chagas disease.
Conflicts of Interest
C.P. and K.M.J. are involved in and receive funding for the development of vaccines against neglected and emerging tropical diseases. The funders had no role in the preparation of this review. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- WHO. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases. Available online: https://apps.who.int/iris/handle/10665/77950 (accessed on 25 March 2024).
- Alarcón de Noya, B.; Jackson, Y. Chagas Disease Epidemiology: From Latin America to the World. In Chagas Disease: A Neglected Tropical Disease; Pinazo Delgado, M.J., Gascón, J., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 27–36. [Google Scholar]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marcondes de Rezende, J. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2012, 26, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Antinori, S.; Galimberti, L.; Bianco, R.; Grande, R.; Galli, M.; Corbellino, M. Chagas disease in Europe: A review for the internist in the globalized world. Eur. J. Intern. Med. 2017, 43, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- CDC. American Trypanosomiasis (Also Known as Chagas Disease). 2021. Available online: https://www.cdc.gov/dpdx/trypanosomiasisamerican/index.html (accessed on 25 March 2024).
- Kirchhoff, L.V. Epidemiology of American Trypanosomiasis (Chagas Disease). Adv. Parasitol. 2011, 75, 1–18. [Google Scholar] [CrossRef]
- Nunes, M.C.P.; Beaton, A.; Acquatella, H.; Bern, C.; Bolger, A.F.; Echeverria, L.E.; Dutra, W.O.; Gascon, J.; Morillo, C.A.; Oliveira-Filho, J.; et al. Chagas Cardiomyopathy: An Update of Current Clinical Knowledge and Management: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e169–e209. [Google Scholar] [CrossRef]
- Nascimento, B.R.; Naback, A.D.N.; Santos, B.M.P.; Geissbühler, Y.; Demacq, C.; Quijano, M.; Perel, P.A.; Molina, I.; Machado, I.E.; Cousin, E.; et al. Prevalence of clinical forms of Chagas disease: A systematic review and meta-analysis—Data from the RAISE study. Lancet Reg. Health Am. 2024, 30, 100681. [Google Scholar] [CrossRef] [PubMed]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public. Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Lascano, F.; Garcia Bournissen, F.; Altcheh, J. Review of pharmacological options for the treatment of Chagas disease. Br. J. Clin. Pharmacol. 2020, 88, 383–402. [Google Scholar] [CrossRef]
- Andrade, M.V.; Noronha, K.; De Souza, A.; Motta-Santos, A.S.; Braga, P.E.F.; Bracarense, H.; De Miranda, M.C.C.; Nascimento, B.R.; Molina, I.; Martins-Melo, F.R.; et al. The economic burden of Chagas disease: A systematic review. PLoS Negl. Trop. Dis. 2023, 17, e0011757. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global economic burden of Chagas disease: A computational simulation model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef]
- Kratz, J.M.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert. Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chagas Disease in Latin America, an Epidemiological Update Based on 2010 Estimates. Available online: https://iris.who.int/bitstream/handle/10665/242316/WER9006_33-44.PDF (accessed on 5 April 2024).
- Bern, C. Antitrypanosomal therapy for chronic Chagas’ disease. N. Engl. J. Med. 2011, 364, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed]
- Viotti, R.; Vigliano, C.; Lococo, B.; Bertocchi, G.; Petti, M.; Alvarez, M.G.; Postan, M.; Armenti, A. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: A nonrandomized trial. Ann. Intern. Med. 2006, 144, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Altcheh, J.; Sierra, V.; Ramirez, T.; Pinto Rocha, J.J.; Grossmann, U.; Huang, E.; Moscatelli, G.; Ding, O. Efficacy and Safety of Nifurtimox in Pediatric Patients with Chagas Disease: Results at 4-Year Follow-Up in a Prospective, Historically Controlled Study (CHICO SECURE). Antimicrob. Agents Chemother. 2023, 67, e0119322. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Vega, C.; Urbina, J.A.; Sanz, S.; Pinazo, M.J.; Pinto, J.J.; Gonzalez, V.R.; Rojas, G.; Ortiz, L.; Garcia, W.; Lozano, D.; et al. New chemotherapy regimens and biomarkers for Chagas disease: The rationale and design of the TESEO study, an open-label, randomised, prospective, phase-2 clinical trial in the Plurinational State of Bolivia. BMJ Open 2021, 11, e052897. [Google Scholar] [CrossRef] [PubMed]
- Cafferata, M.L.; Toscani, M.A.; Althabe, F.; Belizán, J.M.; Bergel, E.; Berrueta, M.; Capparelli, E.V.; Ciganda, Á.; Danesi, E.; Dumonteil, E.; et al. Short-course Benznidazole treatment to reduce Trypanosoma cruzi parasitic load in women of reproductive age (BETTY): A non-inferiority randomized controlled trial study protocol. Reprod. Health 2020, 17, 128. [Google Scholar] [CrossRef]
- Villar, J.C.; Herrera, V.M.; Pérez Carreño, J.G.; Váquiro Herrera, E.; Castellanos Domínguez, Y.Z.; Vásquez, S.M.; Cucunubá, Z.M.; Prado, N.G.; Hernández, Y. Nifurtimox versus benznidazole or placebo for asymptomatic Trypanosoma cruzi infection (Equivalence of Usual Interventions for Trypanosomiasis—EQUITY): Study protocol for a randomised controlled trial. Trials 2019, 20, 431. [Google Scholar] [CrossRef]
- Molina-Morant, D.; Fernandez, M.L.; Bosch-Nicolau, P.; Sulleiro, E.; Bangher, M.; Salvador, F.; Sanchez-Montalva, A.; Ribeiro, A.L.P.; de Paula, A.M.B.; Eloi, S.; et al. Efficacy and safety assessment of different dosage of benznidazol for the treatment of Chagas disease in chronic phase in adults (MULTIBENZ study): Study protocol for a multicenter randomized Phase II non-inferiority clinical trial. Trials 2020, 21, 328. [Google Scholar] [CrossRef]
- Bosch-Nicolau, P.; Fernández, M.L.; Sulleiro, E.; Villar, J.C.; Perez-Molina, J.A.; Correa-Oliveira, R.; Sosa-Estani, S.; Sánchez-Montalvá, A.; Del Carmen Bangher, M.; Moreira, O.C.; et al. Efficacy of three benznidazole dosing strategies for adults living with chronic Chagas disease (MULTIBENZ): An international, randomised, double-blind, phase 2b trial. Lancet Infect. Dis. 2024, 24, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A., Jr.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef]
- Riarte, A.; Prado, N.; Rissio, A.; Velázquez, E.; Ramírez, J.; Vázquez, H.; Tomás, G.; López, S.; Fernández, M.; García, M.; et al. TRAENA: Tratamiento con Benznidazol en pacientes adultos con enfermedad de Chagas crónica de bajo riesgo—Un ensayo clínico aleatorizado en fase 3. Plataforma Investig. Clínica Enferm. Chagas 2016, 4, 6. [Google Scholar]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: Progress and challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef]
- Padilla, A.M.; Wang, W.; Akama, T.; Carter, D.S.; Easom, E.; Freund, Y.; Halladay, J.S.; Liu, Y.; Hamer, S.A.; Hodo, C.L.; et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat. Microbiol. 2022, 7, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, C.E.; Braillard, S.; Glossop, P.A.; Whitlock, G.A.; Jacobs, R.T.; Speake, J.; Pandi, B.; Nare, B.; Maes, L.; Yardley, V.; et al. DNDI-6148: A Novel Benzoxaborole Preclinical Candidate for the Treatment of Visceral Leishmaniasis. J. Med. Chem. 2021, 64, 16159–16176. [Google Scholar] [CrossRef] [PubMed]
- Bahia, M.T.; Andrade, I.M.D.; Martins, T.A.F.; Nascimento, Á.F.D.S.D.; Diniz, L.D.F.; Caldas, I.S.; Talvani, A.; Trunz, B.B.; Torreele, E.; Ribeiro, I. Fexinidazole: A Potential New Drug Candidate for Chagas Disease. PLoS Neglected Trop. Dis. 2012, 6, e1870. [Google Scholar] [CrossRef]
- Diniz, L.D.F.; Mazzeti Ana, L.; Caldas Ivo, S.; Ribeiro, I.; Bahia Maria, T. Outcome of E1224-Benznidazole Combination Treatment for Infection with a Multidrug-Resistant Trypanosoma cruzi Strain in Mice. Antimicrob. Agents Chemother. 2018, 62, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascón, J.; Barreira, F.; Blum, B.; Almeida, I.C.; Alonso-Vega, C.; Barboza, T.; Bilbe, G.; Correia, E.; Garcia, W.; et al. New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): A phase 2, double-blind, randomised trial. Lancet Infect. Dis. 2021, 21, 1129–1140. [Google Scholar] [CrossRef]
- Torrico, F.; Gascon, J.; Ortiz, L.; Alonso-Vega, C.; Pinazo, M.J.; Schijman, A.; Almeida, I.C.; Alves, F.; Strub-Wourgaft, N.; Ribeiro, I. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: A proof-of-concept, randomised, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 419–430. [Google Scholar] [CrossRef]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; Del Carmen Bangher, M.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. Benznidazole and Posaconazole in Eliminating Parasites in Asymptomatic T. cruzi Carriers: The Stop-Chagas Trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascón, J.; Ortiz, L.; Pinto, J.; Rojas, G.; Palacios, A.; Barreira, F.; Blum, B.; Schijman, A.G.; Vaillant, M.; et al. A Phase 2, Randomized, Multicenter, Placebo-Controlled, Proof-of-Concept Trial of Oral Fexinidazole in Adults With Chronic Indeterminate Chagas Disease. Clin. Infect. Dis. 2023, 76, e1186–e1194. [Google Scholar] [CrossRef]
- Dias, J.C.; Silveira, A.C.; Schofield, C.J. The impact of Chagas disease control in Latin America: A review. Mem. Inst. Oswaldo Cruz 2002, 97, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; Stokes-Cawley, O.J.; Buekens, P.; Asti, L.; Bottazzi, M.E.; Strych, U.; Wedlock, P.T.; Mitgang, E.A.; Meymandi, S.; Falcon-Lezama, J.A.; et al. The potential economic value of a therapeutic Chagas disease vaccine for pregnant women to prevent congenital transmission. Vaccine 2020, 38, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.S.; Dutra, W.O.; Esper, L.; Gollob, K.J.; Teixeira, M.M.; Factor, S.M.; Weiss, L.M.; Nagajyothi, F.; Tanowitz, H.B.; Garg, N.J. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol. 2012, 34, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Acosta Rodríguez, E.V.; Araujo Furlan, C.L.; Fiocca Vernengo, F.; Montes, C.L.; Gruppi, A. Understanding CD8(+) T Cell Immunity to Trypanosoma cruzi and How to Improve It. Trends Parasitol. 2019, 35, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Herati, R.S.; Wherry, E.J. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Consideration of Strategies to Improve the Value of Animal Models. Cold Spring Harb. Perspect. Biol. 2018, 10, a031583. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic. Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Pinazo, M.J.; Thomas, M.C.; Bustamante, J.; Almeida, I.C.; Lopez, M.C.; Gascon, J. Biomarkers of therapeutic responses in chronic Chagas disease: State of the art and future perspectives. Mem. Inst. Oswaldo Cruz 2015, 110, 422–432. [Google Scholar] [CrossRef]
- Alpern, J.D.; Lopez-Velez, R.; Stauffer, W.M. Access to benznidazole for Chagas disease in the United States—Cautious optimism? PLoS Neglected Trop. Dis. 2017, 11, e0005794. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. Challenges of Developing Novel Vaccines with Particular Global Health Importance. Front. Immunol. 2020, 11, 517290. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.C.; Plotkin, S.A. Impact of Vaccines; Health, Economic and Social Perspectives. Front. Microbiol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Dumonteil, E.; Bottazzi, M.E.; Zhan, B.; Heffernan, M.J.; Jones, K.; Valenzuela, J.G.; Kamhawi, S.; Ortega, J.; Rosales, S.P.D.L.; Lee, B.Y.; et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: Rationale and prospects. Expert Rev. Vaccines 2012, 11, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Hernandez, I.; Dumonteil, E. Advances and challenges towards a vaccine against Chagas disease. Hum. Vaccines 2011, 7, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.P.; Barrio, A.B.; Cardozo, R.M.; Duffy, T.; Schijman, A.G.; Basombrío, M.A. Impairment of Infectivity and Immunoprotective Effect of a LYT1 Null Mutant of Trypanosoma cruzi. Infect. Immun. 2008, 76, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Basombrío, M.A.; Gómez, L.; Padilla, A.M.; Ciaccio, M.; Nozaki, T.; Cross, G.A.M. Targeted Deletion of the Gp72 Gene Decreases the Infectivity of Trypanosoma Cruzi For Mice And Insect Vectors. J. Parasitol. 2002, 88, 489–493+485. [Google Scholar] [CrossRef] [PubMed]
- Perez Brandan, C.; Padilla, A.M.; Xu, D.; Tarleton, R.L.; Basombrio, M.A. Knockout of the dhfr-ts Gene in Trypanosoma cruzi Generates Attenuated Parasites Able to Confer Protection against a Virulent Challenge. PLoS Neglected Trop. Dis. 2011, 5, e1418. [Google Scholar] [CrossRef] [PubMed]
- Collins Matthew, H.; Craft Julie, M.; Bustamante Juan, M.; Tarleton Rick, L. Oral Exposure to Trypanosoma cruzi Elicits a Systemic CD8+ T Cell Response and Protection against Heterotopic Challenge. Infect. Immun. 2011, 79, 3397–3406. [Google Scholar] [CrossRef]
- Schnapp, A.R.; Eickhoff, C.S.; Scharfstein, J.; Hoft, D.F. Induction of B- and T-cell responses to cruzipain in the murine model of Trypanosoma cruzi infection. Microbes Infect. 2002, 4, 805–813. [Google Scholar] [CrossRef]
- Chou, B.; Hiromatsu, K.; Hisaeda, H.; Duan, X.; Imai, T.; Murata, S.; Tanaka, K.; Himeno, K. Genetic immunization based on the ubiquitin-fusion degradation pathway against Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2010, 392, 277–282. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Ramos-Ligonio, A.; López-Monteón, A.; Salgado-Jiménez, B.; Talamás-Rohana, P.; Rosales-Encina, J.L. A DNA Vaccine Encoding for TcSSP4 Induces Protection against Acute and Chronic Infection in Experimental Chagas Disease. Int. J. Biol. Sci. 2011, 7, 1230–1238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoft, D.F.; Eickhoff, C.S.; Giddings, O.K.; Vasconcelos, J.R.C.; Rodrigues, M.c.M. Trans-Sialidase Recombinant Protein Mixed with CpG Motif-Containing Oligodeoxynucleotide Induces Protective Mucosal and Systemic Trypanosoma cruzi Immunity Involving CD8+ CTL and B Cell-Mediated Cross-Priming1. J. Immunol. 2007, 179, 6889–6900. [Google Scholar] [CrossRef] [PubMed]
- Giddings, O.K.; Eickhoff, C.S.; Sullivan, N.L.; Hoft, D.F. Intranasal Vaccinations with the trans-Sialidase Antigen plus CpG Adjuvant Induce Mucosal Immunity Protective against Conjunctival Trypanosoma cruzi Challenges. Infect. Immun. 2010, 78, 1333–1338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eickhoff, C.S.; Vasconcelos, J.R.; Sullivan, N.L.; Blazevic, A.; Bruna-Romero, O.; Rodrigues, M.M.; Hoft, D.F. Co-Administration of a Plasmid DNA Encoding IL-15 Improves Long-Term Protection of a Genetic Vaccine against Trypanosoma cruzi. PLoS Neglected Trop. Dis. 2011, 5, e983. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, Y.; Takashima, Y.; Kobayashi, S.; Matsumoto, Y.; Takeuchi, T.; Ohyanagi-Hara, M.; Yoshida, A.; Ohwada, A.; Akiba, H.; Yagita, H.; et al. Immune Responses against a Single CD8+-T-Cell Epitope Induced by Virus Vector Vaccination Can Successfully Control Trypanosoma cruzi Infection. Infect. Immun. 2005, 73, 7356–7365. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.V.; Cardoso, J.E.; Claser, C.; Rodrigues, M.M.; Gazzinelli, R.T.; Bruna-Romero, O. Long-term protective immunity induced against Trypanosoma cruzi infection after vaccination with recombinant adenoviruses encoding amastigote surface protein-2 and trans-sialidase. Hum. Gene Ther. 2006, 17, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Parodi, C.; Padilla, A.M.; Basombrío, M.A. Protective immunity against Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2009, 104, 288–294. [Google Scholar] [CrossRef]
- Cazorla, S.I.; Frank, F.M.; Malchiodi, E.L. Vaccination approaches against Trypanosoma cruzi infection. Expert. Rev. Vaccines 2009, 8, 921–935. [Google Scholar] [CrossRef]
- De Alencar, B.C.; Araújo, A.F.; Penido, M.L.; Gazzinelli, R.T.; Rodrigues, M.M. Cross-priming of long lived protective CD8+ T cells against Trypanosoma cruzi infection: Importance of a TLR9 agonist and CD4+ T cells. Vaccine 2007, 25, 6018–6027. [Google Scholar] [CrossRef]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Arnaiz, M.; Mirkin, G.A.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Redirection of the Immune Response to the Functional Catalytic Domain of the Cystein Proteinase Cruzipain Improves Protective Immunity against Trypanosoma cruzi Infection. J. Infect. Dis. 2010, 202, 136–144. [Google Scholar] [CrossRef]
- Frank, F.M.; Petray, P.B.; Cazorla, S.I.; Muñoz, M.C.; Corral, R.S.; Malchiodi, E.L. Use of a purified Trypanosoma cruzi antigen and CpG oligodeoxynucleotides for immunoprotection against a lethal challenge with trypomastigotes. Vaccine 2003, 22, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, C.S.; Giddings, O.K.; Yoshida, N.; Hoft, D.F. Immune responses to gp82 provide protection against mucosal Trypanosoma cruzi infection. Mem. Inst. Oswaldo Cruz 2010, 105, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Serna, C.; Lara, J.A.; Rodrigues, S.P.; Marques, A.F.; Almeida, I.C.; Maldonado, R.A. A synthetic peptide from Trypanosoma cruzi mucin-like associated surface protein as candidate for a vaccine against Chagas disease. Vaccine 2014, 32, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Campos, V.; Martinez-Vega, P.; Ramirez-Sierra, M.J.; Rosado-Vallado, M.; Seid, C.A.; Hudspeth, E.M.; Wei, J.; Liu, Z.; Kwityn, C.; Hammond, M.; et al. Expression, purification, immunogenicity, and protective efficacy of a recombinant Tc24 antigen as a vaccine against Trypanosoma cruzi infection in mice. Vaccine 2015, 33, 4505–4512. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.A.; Versteeg, L.; Wang, Q.; Pollet, J.; Zhan, B.; Gusovsky, F.; Bottazzi, M.E.; Hotez, P.J.; Jones, K.M. A therapeutic vaccine prototype induces protective immunity and reduces cardiac fibrosis in a mouse model of chronic Trypanosoma cruzi infection. PLoS Neglected Trop. Dis. 2019, 13, e0007413. [Google Scholar] [CrossRef] [PubMed]
- Rios, L.E.; Vázquez-Chagoyán, J.C.; Pacheco, A.O.; Zago, M.P.; Garg, N.J. Immunity and vaccine development efforts against Trypanosoma cruzi. Acta Tropica 2019, 200, 105168. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.R.; Almeida, I.C. Carbohydrate immunity in American trypanosomiasis. Springer Semin. Immunopathol. 1993, 15, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Almeida, I.C.; Milani, S.R.; Gorin, P.A.; Travassos, L.R. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-alpha-galactosyl antibodies. J. Immunol. 1991, 146, 2394–2400. [Google Scholar] [CrossRef]
- Almeida, I.C.; Ferguson, M.A.; Schenkman, S.; Travassos, L.R. Lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem. J. 1994, 304, 793–802. [Google Scholar] [CrossRef]
- Almeida, I.C.; Krautz, G.M.; Krettli, A.U.; Travassos, L.R. Glycoconjugates of Trypanosoma cruzi: A 74 kD antigen of trypomastigotes specifically reacts with lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas disease. J. Clin. Lab. Anal. 1993, 7, 307–316. [Google Scholar] [CrossRef]
- Portillo, S.; Zepeda, B.G.; Iniguez, E.; Olivas, J.J.; Karimi, N.H.; Moreira, O.C.; Marques, A.F.; Michael, K.; Maldonado, R.A.; Almeida, I.C. A prophylactic alpha-Gal-based glycovaccine effectively protects against murine acute Chagas disease. NPJ Vaccines 2019, 4, 13. [Google Scholar] [CrossRef]
- Iniguez, E.; Schocker, N.S.; Subramaniam, K.; Portillo, S.; Montoya, A.L.; Al-Salem, W.S.; Torres, C.L.; Rodriguez, F.; Moreira, O.C.; Acosta-Serrano, A.; et al. An alpha-Gal-containing neoglycoprotein-based vaccine partially protects against murine cutaneous leishmaniasis caused by Leishmania major. PLoS Negl. Trop. Dis. 2017, 11, e0006039. [Google Scholar] [CrossRef] [PubMed]
- Parussini, F.; Duschak, V.G.; Cazzulo, J.J. Membrane-bound cysteine proteinase isoforms in different developmental stages of Trypanosoma cruzi. Cell Mol. Biol. 1998, 44, 513–519. [Google Scholar]
- Bhatia, V.; Sinha, M.; Luxon, B.; Garg, N. Utility of the Trypanosoma cruzi Sequence Database for Identification of Potential Vaccine Candidates by In Silico and In Vitro Screening. Infect. Immun. 2004, 72, 6245–6254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garg, N.; Tarleton Rick, L. Genetic Immunization Elicits Antigen-Specific Protective Immune Responses and Decreases Disease Severity in Trypanosoma cruzi Infection. Infect. Immun. 2002, 70, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Albareda, M.C.; Natale, M.A.; De Rissio, A.M.; Fernandez, M.; Serjan, A.; Alvarez, M.G.; Cooley, G.; Shen, H.; Viotti, R.; Bua, J.; et al. Distinct Treatment Outcomes of Antiparasitic Therapy in Trypanosoma cruzi-Infected Children Is Associated with Early Changes in Cytokines, Chemokines, and T-Cell Phenotypes. Front. Immunol. 2018, 9, 1958. [Google Scholar] [CrossRef]
- Limon-Flores, A.Y.; Cervera-Cetina, R.; Tzec-Arjona, J.L.; Ek-Macias, L.; Sánchez-Burgos, G.; Ramirez-Sierra, M.J.; Cruz-Chan, J.V.; VanWynsberghe, N.R.; Dumonteil, E. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice: Role of CD4+ and CD8+ T cells. Vaccine 2010, 28, 7414–7419. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Scientific Reports 2017, 7, 252. [Google Scholar] [CrossRef]
- Sanchez Alberti, A.; Bivona, A.E.; Matos, M.N.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; González, G.; Morales, C.; Cardoso, A.C.; et al. Mucosal Heterologous Prime/Boost Vaccination Induces Polyfunctional Systemic Immunity, Improving Protection Against Trypanosoma cruzi. Front. Immunol. 2020, 11, 128. [Google Scholar] [CrossRef]
- Araújo, A.F.S.; De Alencar, B.C.G.; Vasconcelos, J.R.C.; Hiyane, M.I.; Marinho, C.R.F.; Penido, M.L.O.; Boscardin, S.B.; Hoft, D.F.; Gazzinelli, R.T.; Rodrigues, M.M. CD8+-T-Cell-Dependent Control of Trypanosoma cruzi Infection in a Highly Susceptible Mouse Strain after Immunization with Recombinant Proteins Based on Amastigote Surface Protein 2. Infect. Immun. 2005, 73, 6017–6025. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Garg, N.J. Prophylactic Efficacy of TcVac2 against Trypanosoma cruzi in Mice. PLoS Neglected Trop. Dis. 2010, 4, e797. [Google Scholar] [CrossRef] [PubMed]
- Rigato, P.O.; De Alencar, B.C.; De Vasconcelos, J.R.C.; Dominguez, M.R.; Araújo, A.F.; Machado, A.V.; Gazzinelli, R.T.; Bruna-Romero, O.; Rodrigues, M.M. Heterologous Plasmid DNA Prime-Recombinant Human Adenovirus 5 Boost Vaccination Generates a Stable Pool of Protective Long-Lived CD8+ T Effector Memory Cells Specific for a Human Parasite, Trypanosoma cruzi. Infect. Immun. 2011, 79, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Mancino, C.; Pollet, J.; Zinger, A.; Jones, K.M.; Villar, M.J.; Leao, A.C.; Adhikari, R.; Versteeg, L.; Tyagi Kundu, R.; Strych, U.; et al. Harnessing RNA Technology to Advance Therapeutic Vaccine Antigens against Chagas Disease. ACS Appl. Mater. Interfaces 2024, 16, 15832–15846. [Google Scholar] [CrossRef] [PubMed]
- Poveda, C.; Leão, A.C.; Mancino, C.; Taraballi, F.; Chen, Y.-L.; Adhikari, R.; Villar, M.J.; Kundu, R.; Nguyen, D.M.; Versteeg, L.; et al. Heterologous mRNA-protein vaccination with Tc24 induces a robust cellular immune response against Trypanosoma cruzi, characterized by an increased level of polyfunctional CD8+ T-cells. Curr. Res. Immunol. 2023, 4, 100066. [Google Scholar] [CrossRef]
- Hunter, D.J.; Abdool Karim, S.S.; Baden, L.R.; Farrar, J.J.; Hamel, M.B.; Longo, D.L.; Morrissey, S.; Rubin, E.J. Addressing Vaccine Inequity—Covid-19 Vaccines as a Global Public Good. N. Engl. J. Med. 2022, 386, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- WHO. Vaccine Inequity Undermining Global Economic Recovery. 2021. Available online: https://www.who.int/news/item/22-07-2021-vaccine-inequity-undermining-global-economic-recovery#:~:text=New%20Global%20Dashboard%20on%20COVID,rate%20as%20high%2Dincome%20countries (accessed on 25 March 2024).
- Jones, K.; Versteeg, L.; Damania, A.; Keegan, B.; Kendricks, A.; Pollet, J.; Cruz-Chan Julio, V.; Gusovsky, F.; Hotez Peter, J.; Bottazzi Maria, E. Vaccine-Linked Chemotherapy Improves Benznidazole Efficacy for Acute Chagas Disease. Infect. Immun. 2018, 86, e00876-17. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Chan, J.V.; Villanueva-Lizama, L.E.; Versteeg, L.; Damania, A.; Villar, M.J.; González-López, C.; Keegan, B.; Pollet, J.; Gusovsky, F.; Hotez, P.J.; et al. Vaccine-linked chemotherapy induces IL-17 production and reduces cardiac pathology during acute Trypanosoma cruzi infection. Sci. Rep. 2021, 11, 3222. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.M.; Mangin, E.N.; Reynolds, C.L.; Villanueva, L.E.; Cruz, J.V.; Versteeg, L.; Keegan, B.; Kendricks, A.; Pollet, J.; Gusovsky, F.; et al. Vaccine-linked chemotherapy improves cardiac structure and function in a mouse model of chronic Chagas disease. Front. Cell Infect. Microbiol. 2023, 13, 1106315. [Google Scholar] [CrossRef]
- Liu, Z.; Ulrich vonBargen, R.; Kendricks, A.L.; Wheeler, K.; Leão, A.C.; Sankaranarayanan, K.; Dean, D.A.; Kane, S.S.; Hossain, E.; Pollet, J.; et al. Localized cardiac small molecule trajectories and persistent chemical sequelae in experimental Chagas disease. Nat. Commun. 2023, 14, 6769. [Google Scholar] [CrossRef]
- Prochetto, E.; Bontempi, I.; Rodeles, L.; Cabrera, G.; Vicco, M.; Cacik, P.; Pacini, M.F.; Pérez Gianeselli, M.; Pérez, A.R.; Marcipar, I. Assessment of a combined treatment with a therapeutic vaccine and benznidazole for the Trypanosoma cruzi chronic infection. Acta Trop. 2022, 229, 106334. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, R.B.; Bocchi, E.A. Shorter treatment in chronic Chagas disease: A new promise? Lancet Infect. Dis. 2024, 24, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.F.; Jayawardhana, S.; Lewis, M.D.; White, K.L.; Shackleford, D.M.; Chen, G.; Saunders, J.; Osuna-Cabello, M.; Read, K.D.; Charman, S.A.; et al. Nitroheterocyclic drugs cure experimental Trypanosoma cruzi infections more effectively in the chronic stage than in the acute stage. Sci. Rep. 2016, 6, 35351. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Liu, X.; Stinson, M.; Rivera, I.; Groessl, T.; Tuntland, T.; Yeh, V.; Wen, B.; Molteni, V.; Glynne, R.; et al. Antitrypanosomal Treatment with Benznidazole Is Superior to Posaconazole Regimens in Mouse Models of Chagas Disease. Antimicrob. Agents Chemother. 2015, 59, 6385–6394. [Google Scholar] [CrossRef]
- Branquinho, R.T.; Mosqueira, V.C.; De Oliveira-Silva, J.C.; Simões-Silva, M.R.; Saúde-Guimarães, D.A.; De Lana, M. Sesquiterpene lactone in nanostructured parenteral dosage form is efficacious in experimental Chagas disease. Antimicrob. Agents Chemother. 2014, 58, 2067–2075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Mello, C.G.; Branquinho, R.T.; Oliveira, M.T.; Milagre, M.M.; Saúde-Guimarães, D.A.; Mosqueira, V.C.; Lana, M. Efficacy of Lychnopholide Polymeric Nanocapsules after Oral and Intravenous Administration in Murine Experimental Chagas Disease. Antimicrob. Agents Chemother. 2016, 60, 5215–5222. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Capelari-Oliveira, P.; Bahia, M.T.; Mosqueira, V.C.F. Review on Experimental Treatment Strategies Against Trypanosoma cruzi. J. Exp. Pharmacol. 2021, 13, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.P.; Gazzinelli, R.T.; Morel, C.M.; Precioso, A.R. Why do we still have not a vaccine against Chagas disease? Mem. Inst. Oswaldo Cruz 2022, 117, e200314. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Part 2: Potential Alternatives to the Use of Animals in Preclinical Trials. JACC Basic. Transl. Sci. 2020, 5, 387–397. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C. The Case for the Development of a Chagas Disease Vaccine: Why? How? When? Trop. Med. Infect. Dis. 2021, 6, 16. [Google Scholar] [CrossRef]
- Brown, C.M.; Slavinski, S.; Ettestad, P.; Sidwa, T.J.; Sorhage, F.E. Compendium of Animal Rabies Prevention and Control, 2016. J. Am. Vet. Med. Assoc. 2016, 248, 505–517. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).