
Treatments and the Perspectives of Developing a Vaccine for Chagas Disease


Abstract
:1. Introduction
2. Treatment
2.1. Current Treatment
2.2. New Treatments
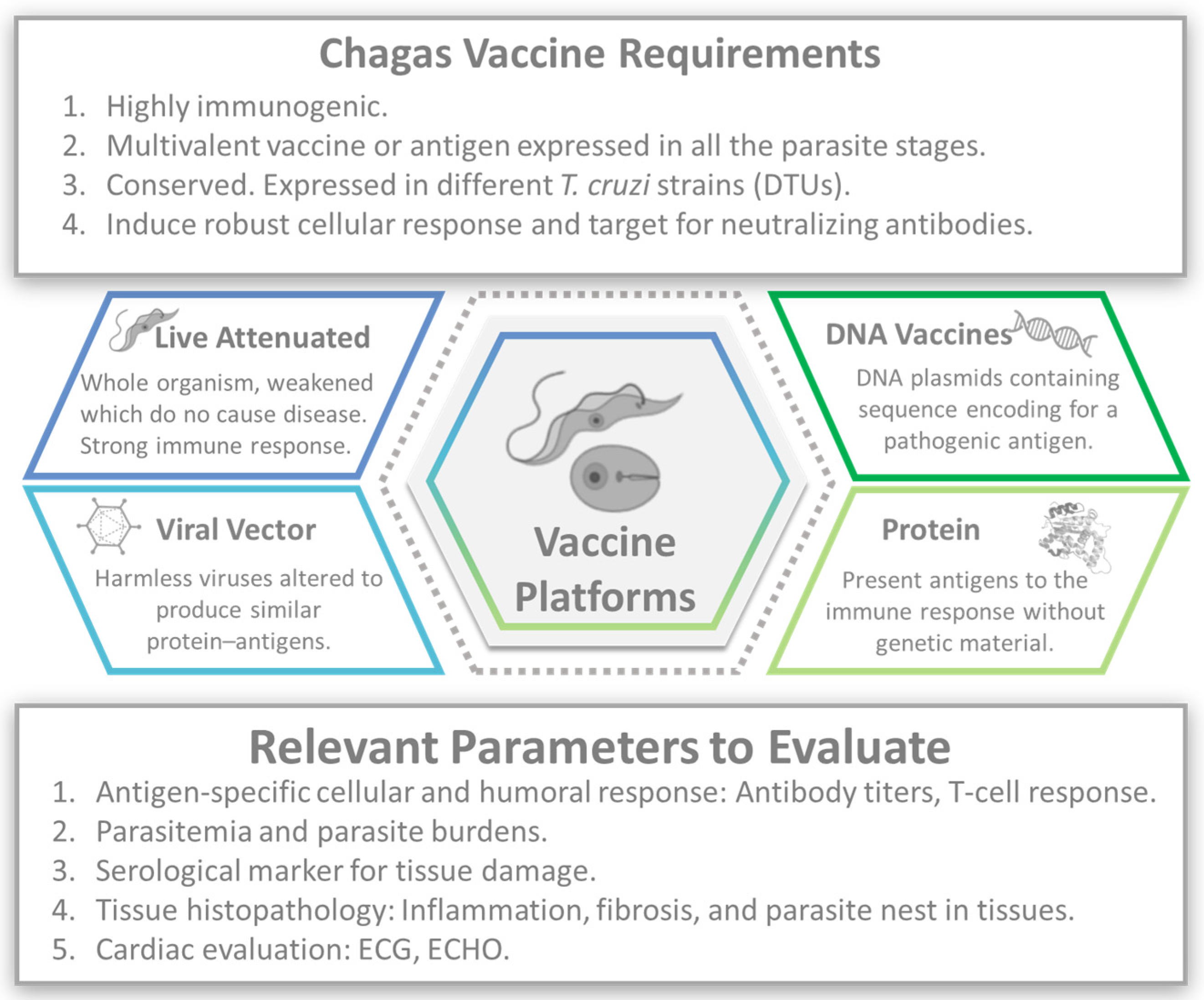
3. Vaccines
- Complex parasite life cycle: T. cruzi, has a complex life cycle involving different stages and multiple forms of the parasite, and each stage may require a different type of immune response for effective control and eradication [6].
- Limited understanding of protective immunity: The precise immune response required to confer protection against CD is not fully understood yet [40,41]. Identifying the key immune mechanisms involved in controlling the infection and developing vaccines that elicit those specific responses is essential to creating an effective CD vaccine.
- Lack of surrogate markers of protection: To date, no established surrogate markers can reliably predict vaccine efficacy against CD [44]. The absence of such markers makes it challenging to assess the efficacy of vaccine candidates in clinical trials and may require long-term follow-up to determine their effectiveness.
- Limited commercial interest: CD primarily affects marginalized and economically disadvantaged populations, predominantly in Latin America. The lack of financial incentives for pharmaceutical companies has historically hindered the development of vaccines for neglected tropical diseases. Public–private partnerships and alternative funding mechanisms might be necessary to overcome this challenge [45].
- Regulatory and manufacturing challenges: Vaccine development involves navigating complex regulatory processes and scaling production to meet global demand [46]. Regulatory approvals, manufacturing infrastructure, and cost-effectiveness are significant challenges that must be addressed to ensure access to an affordable and widely available CD vaccine [45].
3.1. Attenuated Vaccines
3.2. DNA Vaccines
3.3. Viral Vector
3.4. Recombinant Protein or Peptide Vaccines
3.5. Glycoconjugates
3.6. Multivalent Vaccines
3.7. Heterologous Vaccines
3.8. mRNA Vaccines
4. Vaccine-Linked Chemotherapy
5. Discussion
6. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases. Available online: https://apps.who.int/iris/handle/10665/77950 (accessed on 25 March 2024).
- Alarcón de Noya, B.; Jackson, Y. Chagas Disease Epidemiology: From Latin America to the World. In Chagas Disease: A Neglected Tropical Disease; Pinazo Delgado, M.J., Gascón, J., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 27–36. [Google Scholar]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marcondes de Rezende, J. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2012, 26, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Antinori, S.; Galimberti, L.; Bianco, R.; Grande, R.; Galli, M.; Corbellino, M. Chagas disease in Europe: A review for the internist in the globalized world. Eur. J. Intern. Med. 2017, 43, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- CDC. American Trypanosomiasis (Also Known as Chagas Disease). 2021. Available online: https://www.cdc.gov/dpdx/trypanosomiasisamerican/index.html (accessed on 25 March 2024).
- Kirchhoff, L.V. Epidemiology of American Trypanosomiasis (Chagas Disease). Adv. Parasitol. 2011, 75, 1–18. [Google Scholar] [CrossRef]
- Nunes, M.C.P.; Beaton, A.; Acquatella, H.; Bern, C.; Bolger, A.F.; Echeverria, L.E.; Dutra, W.O.; Gascon, J.; Morillo, C.A.; Oliveira-Filho, J.; et al. Chagas Cardiomyopathy: An Update of Current Clinical Knowledge and Management: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e169–e209. [Google Scholar] [CrossRef]
- Nascimento, B.R.; Naback, A.D.N.; Santos, B.M.P.; Geissbühler, Y.; Demacq, C.; Quijano, M.; Perel, P.A.; Molina, I.; Machado, I.E.; Cousin, E.; et al. Prevalence of clinical forms of Chagas disease: A systematic review and meta-analysis—Data from the RAISE study. Lancet Reg. Health Am. 2024, 30, 100681. [Google Scholar] [CrossRef] [PubMed]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public. Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Lascano, F.; Garcia Bournissen, F.; Altcheh, J. Review of pharmacological options for the treatment of Chagas disease. Br. J. Clin. Pharmacol. 2020, 88, 383–402. [Google Scholar] [CrossRef]
- Andrade, M.V.; Noronha, K.; De Souza, A.; Motta-Santos, A.S.; Braga, P.E.F.; Bracarense, H.; De Miranda, M.C.C.; Nascimento, B.R.; Molina, I.; Martins-Melo, F.R.; et al. The economic burden of Chagas disease: A systematic review. PLoS Negl. Trop. Dis. 2023, 17, e0011757. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global economic burden of Chagas disease: A computational simulation model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef]
- Kratz, J.M.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert. Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chagas Disease in Latin America, an Epidemiological Update Based on 2010 Estimates. Available online: https://iris.who.int/bitstream/handle/10665/242316/WER9006_33-44.PDF (accessed on 5 April 2024).
- Bern, C. Antitrypanosomal therapy for chronic Chagas’ disease. N. Engl. J. Med. 2011, 364, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed]
- Viotti, R.; Vigliano, C.; Lococo, B.; Bertocchi, G.; Petti, M.; Alvarez, M.G.; Postan, M.; Armenti, A. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: A nonrandomized trial. Ann. Intern. Med. 2006, 144, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Altcheh, J.; Sierra, V.; Ramirez, T.; Pinto Rocha, J.J.; Grossmann, U.; Huang, E.; Moscatelli, G.; Ding, O. Efficacy and Safety of Nifurtimox in Pediatric Patients with Chagas Disease: Results at 4-Year Follow-Up in a Prospective, Historically Controlled Study (CHICO SECURE). Antimicrob. Agents Chemother. 2023, 67, e0119322. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Vega, C.; Urbina, J.A.; Sanz, S.; Pinazo, M.J.; Pinto, J.J.; Gonzalez, V.R.; Rojas, G.; Ortiz, L.; Garcia, W.; Lozano, D.; et al. New chemotherapy regimens and biomarkers for Chagas disease: The rationale and design of the TESEO study, an open-label, randomised, prospective, phase-2 clinical trial in the Plurinational State of Bolivia. BMJ Open 2021, 11, e052897. [Google Scholar] [CrossRef] [PubMed]
- Cafferata, M.L.; Toscani, M.A.; Althabe, F.; Belizán, J.M.; Bergel, E.; Berrueta, M.; Capparelli, E.V.; Ciganda, Á.; Danesi, E.; Dumonteil, E.; et al. Short-course Benznidazole treatment to reduce Trypanosoma cruzi parasitic load in women of reproductive age (BETTY): A non-inferiority randomized controlled trial study protocol. Reprod. Health 2020, 17, 128. [Google Scholar] [CrossRef]
- Villar, J.C.; Herrera, V.M.; Pérez Carreño, J.G.; Váquiro Herrera, E.; Castellanos Domínguez, Y.Z.; Vásquez, S.M.; Cucunubá, Z.M.; Prado, N.G.; Hernández, Y. Nifurtimox versus benznidazole or placebo for asymptomatic Trypanosoma cruzi infection (Equivalence of Usual Interventions for Trypanosomiasis—EQUITY): Study protocol for a randomised controlled trial. Trials 2019, 20, 431. [Google Scholar] [CrossRef]
- Molina-Morant, D.; Fernandez, M.L.; Bosch-Nicolau, P.; Sulleiro, E.; Bangher, M.; Salvador, F.; Sanchez-Montalva, A.; Ribeiro, A.L.P.; de Paula, A.M.B.; Eloi, S.; et al. Efficacy and safety assessment of different dosage of benznidazol for the treatment of Chagas disease in chronic phase in adults (MULTIBENZ study): Study protocol for a multicenter randomized Phase II non-inferiority clinical trial. Trials 2020, 21, 328. [Google Scholar] [CrossRef]
- Bosch-Nicolau, P.; Fernández, M.L.; Sulleiro, E.; Villar, J.C.; Perez-Molina, J.A.; Correa-Oliveira, R.; Sosa-Estani, S.; Sánchez-Montalvá, A.; Del Carmen Bangher, M.; Moreira, O.C.; et al. Efficacy of three benznidazole dosing strategies for adults living with chronic Chagas disease (MULTIBENZ): An international, randomised, double-blind, phase 2b trial. Lancet Infect. Dis. 2024, 24, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A., Jr.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef]
- Riarte, A.; Prado, N.; Rissio, A.; Velázquez, E.; Ramírez, J.; Vázquez, H.; Tomás, G.; López, S.; Fernández, M.; García, M.; et al. TRAENA: Tratamiento con Benznidazol en pacientes adultos con enfermedad de Chagas crónica de bajo riesgo—Un ensayo clínico aleatorizado en fase 3. Plataforma Investig. Clínica Enferm. Chagas 2016, 4, 6. [Google Scholar]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: Progress and challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef]
- Padilla, A.M.; Wang, W.; Akama, T.; Carter, D.S.; Easom, E.; Freund, Y.; Halladay, J.S.; Liu, Y.; Hamer, S.A.; Hodo, C.L.; et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat. Microbiol. 2022, 7, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, C.E.; Braillard, S.; Glossop, P.A.; Whitlock, G.A.; Jacobs, R.T.; Speake, J.; Pandi, B.; Nare, B.; Maes, L.; Yardley, V.; et al. DNDI-6148: A Novel Benzoxaborole Preclinical Candidate for the Treatment of Visceral Leishmaniasis. J. Med. Chem. 2021, 64, 16159–16176. [Google Scholar] [CrossRef] [PubMed]
- Bahia, M.T.; Andrade, I.M.D.; Martins, T.A.F.; Nascimento, Á.F.D.S.D.; Diniz, L.D.F.; Caldas, I.S.; Talvani, A.; Trunz, B.B.; Torreele, E.; Ribeiro, I. Fexinidazole: A Potential New Drug Candidate for Chagas Disease. PLoS Neglected Trop. Dis. 2012, 6, e1870. [Google Scholar] [CrossRef]
- Diniz, L.D.F.; Mazzeti Ana, L.; Caldas Ivo, S.; Ribeiro, I.; Bahia Maria, T. Outcome of E1224-Benznidazole Combination Treatment for Infection with a Multidrug-Resistant Trypanosoma cruzi Strain in Mice. Antimicrob. Agents Chemother. 2018, 62, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascón, J.; Barreira, F.; Blum, B.; Almeida, I.C.; Alonso-Vega, C.; Barboza, T.; Bilbe, G.; Correia, E.; Garcia, W.; et al. New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): A phase 2, double-blind, randomised trial. Lancet Infect. Dis. 2021, 21, 1129–1140. [Google Scholar] [CrossRef]
- Torrico, F.; Gascon, J.; Ortiz, L.; Alonso-Vega, C.; Pinazo, M.J.; Schijman, A.; Almeida, I.C.; Alves, F.; Strub-Wourgaft, N.; Ribeiro, I. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: A proof-of-concept, randomised, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 419–430. [Google Scholar] [CrossRef]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; Del Carmen Bangher, M.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. Benznidazole and Posaconazole in Eliminating Parasites in Asymptomatic T. cruzi Carriers: The Stop-Chagas Trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascón, J.; Ortiz, L.; Pinto, J.; Rojas, G.; Palacios, A.; Barreira, F.; Blum, B.; Schijman, A.G.; Vaillant, M.; et al. A Phase 2, Randomized, Multicenter, Placebo-Controlled, Proof-of-Concept Trial of Oral Fexinidazole in Adults With Chronic Indeterminate Chagas Disease. Clin. Infect. Dis. 2023, 76, e1186–e1194. [Google Scholar] [CrossRef]
- Dias, J.C.; Silveira, A.C.; Schofield, C.J. The impact of Chagas disease control in Latin America: A review. Mem. Inst. Oswaldo Cruz 2002, 97, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; Stokes-Cawley, O.J.; Buekens, P.; Asti, L.; Bottazzi, M.E.; Strych, U.; Wedlock, P.T.; Mitgang, E.A.; Meymandi, S.; Falcon-Lezama, J.A.; et al. The potential economic value of a therapeutic Chagas disease vaccine for pregnant women to prevent congenital transmission. Vaccine 2020, 38, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.S.; Dutra, W.O.; Esper, L.; Gollob, K.J.; Teixeira, M.M.; Factor, S.M.; Weiss, L.M.; Nagajyothi, F.; Tanowitz, H.B.; Garg, N.J. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol. 2012, 34, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Acosta Rodríguez, E.V.; Araujo Furlan, C.L.; Fiocca Vernengo, F.; Montes, C.L.; Gruppi, A. Understanding CD8(+) T Cell Immunity to Trypanosoma cruzi and How to Improve It. Trends Parasitol. 2019, 35, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Herati, R.S.; Wherry, E.J. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Consideration of Strategies to Improve the Value of Animal Models. Cold Spring Harb. Perspect. Biol. 2018, 10, a031583. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic. Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Pinazo, M.J.; Thomas, M.C.; Bustamante, J.; Almeida, I.C.; Lopez, M.C.; Gascon, J. Biomarkers of therapeutic responses in chronic Chagas disease: State of the art and future perspectives. Mem. Inst. Oswaldo Cruz 2015, 110, 422–432. [Google Scholar] [CrossRef]
- Alpern, J.D.; Lopez-Velez, R.; Stauffer, W.M. Access to benznidazole for Chagas disease in the United States—Cautious optimism? PLoS Neglected Trop. Dis. 2017, 11, e0005794. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. Challenges of Developing Novel Vaccines with Particular Global Health Importance. Front. Immunol. 2020, 11, 517290. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.C.; Plotkin, S.A. Impact of Vaccines; Health, Economic and Social Perspectives. Front. Microbiol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Dumonteil, E.; Bottazzi, M.E.; Zhan, B.; Heffernan, M.J.; Jones, K.; Valenzuela, J.G.; Kamhawi, S.; Ortega, J.; Rosales, S.P.D.L.; Lee, B.Y.; et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: Rationale and prospects. Expert Rev. Vaccines 2012, 11, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Hernandez, I.; Dumonteil, E. Advances and challenges towards a vaccine against Chagas disease. Hum. Vaccines 2011, 7, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.P.; Barrio, A.B.; Cardozo, R.M.; Duffy, T.; Schijman, A.G.; Basombrío, M.A. Impairment of Infectivity and Immunoprotective Effect of a LYT1 Null Mutant of Trypanosoma cruzi. Infect. Immun. 2008, 76, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Basombrío, M.A.; Gómez, L.; Padilla, A.M.; Ciaccio, M.; Nozaki, T.; Cross, G.A.M. Targeted Deletion of the Gp72 Gene Decreases the Infectivity of Trypanosoma Cruzi For Mice And Insect Vectors. J. Parasitol. 2002, 88, 489–493+485. [Google Scholar] [CrossRef] [PubMed]
- Perez Brandan, C.; Padilla, A.M.; Xu, D.; Tarleton, R.L.; Basombrio, M.A. Knockout of the dhfr-ts Gene in Trypanosoma cruzi Generates Attenuated Parasites Able to Confer Protection against a Virulent Challenge. PLoS Neglected Trop. Dis. 2011, 5, e1418. [Google Scholar] [CrossRef] [PubMed]
- Collins Matthew, H.; Craft Julie, M.; Bustamante Juan, M.; Tarleton Rick, L. Oral Exposure to Trypanosoma cruzi Elicits a Systemic CD8+ T Cell Response and Protection against Heterotopic Challenge. Infect. Immun. 2011, 79, 3397–3406. [Google Scholar] [CrossRef]
- Schnapp, A.R.; Eickhoff, C.S.; Scharfstein, J.; Hoft, D.F. Induction of B- and T-cell responses to cruzipain in the murine model of Trypanosoma cruzi infection. Microbes Infect. 2002, 4, 805–813. [Google Scholar] [CrossRef]
- Chou, B.; Hiromatsu, K.; Hisaeda, H.; Duan, X.; Imai, T.; Murata, S.; Tanaka, K.; Himeno, K. Genetic immunization based on the ubiquitin-fusion degradation pathway against Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2010, 392, 277–282. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Ramos-Ligonio, A.; López-Monteón, A.; Salgado-Jiménez, B.; Talamás-Rohana, P.; Rosales-Encina, J.L. A DNA Vaccine Encoding for TcSSP4 Induces Protection against Acute and Chronic Infection in Experimental Chagas Disease. Int. J. Biol. Sci. 2011, 7, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Hoft, D.F.; Eickhoff, C.S.; Giddings, O.K.; Vasconcelos, J.R.C.; Rodrigues, M.c.M. Trans-Sialidase Recombinant Protein Mixed with CpG Motif-Containing Oligodeoxynucleotide Induces Protective Mucosal and Systemic Trypanosoma cruzi Immunity Involving CD8+ CTL and B Cell-Mediated Cross-Priming1. J. Immunol. 2007, 179, 6889–6900. [Google Scholar] [CrossRef] [PubMed]
- Giddings, O.K.; Eickhoff, C.S.; Sullivan, N.L.; Hoft, D.F. Intranasal Vaccinations with the trans-Sialidase Antigen plus CpG Adjuvant Induce Mucosal Immunity Protective against Conjunctival Trypanosoma cruzi Challenges. Infect. Immun. 2010, 78, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, C.S.; Vasconcelos, J.R.; Sullivan, N.L.; Blazevic, A.; Bruna-Romero, O.; Rodrigues, M.M.; Hoft, D.F. Co-Administration of a Plasmid DNA Encoding IL-15 Improves Long-Term Protection of a Genetic Vaccine against Trypanosoma cruzi. PLoS Neglected Trop. Dis. 2011, 5, e983. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, Y.; Takashima, Y.; Kobayashi, S.; Matsumoto, Y.; Takeuchi, T.; Ohyanagi-Hara, M.; Yoshida, A.; Ohwada, A.; Akiba, H.; Yagita, H.; et al. Immune Responses against a Single CD8+-T-Cell Epitope Induced by Virus Vector Vaccination Can Successfully Control Trypanosoma cruzi Infection. Infect. Immun. 2005, 73, 7356–7365. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.V.; Cardoso, J.E.; Claser, C.; Rodrigues, M.M.; Gazzinelli, R.T.; Bruna-Romero, O. Long-term protective immunity induced against Trypanosoma cruzi infection after vaccination with recombinant adenoviruses encoding amastigote surface protein-2 and trans-sialidase. Hum. Gene Ther. 2006, 17, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Parodi, C.; Padilla, A.M.; Basombrío, M.A. Protective immunity against Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2009, 104, 288–294. [Google Scholar] [CrossRef]
- Cazorla, S.I.; Frank, F.M.; Malchiodi, E.L. Vaccination approaches against Trypanosoma cruzi infection. Expert. Rev. Vaccines 2009, 8, 921–935. [Google Scholar] [CrossRef]
- De Alencar, B.C.; Araújo, A.F.; Penido, M.L.; Gazzinelli, R.T.; Rodrigues, M.M. Cross-priming of long lived protective CD8+ T cells against Trypanosoma cruzi infection: Importance of a TLR9 agonist and CD4+ T cells. Vaccine 2007, 25, 6018–6027. [Google Scholar] [CrossRef]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Arnaiz, M.; Mirkin, G.A.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Redirection of the Immune Response to the Functional Catalytic Domain of the Cystein Proteinase Cruzipain Improves Protective Immunity against Trypanosoma cruzi Infection. J. Infect. Dis. 2010, 202, 136–144. [Google Scholar] [CrossRef]
- Frank, F.M.; Petray, P.B.; Cazorla, S.I.; Muñoz, M.C.; Corral, R.S.; Malchiodi, E.L. Use of a purified Trypanosoma cruzi antigen and CpG oligodeoxynucleotides for immunoprotection against a lethal challenge with trypomastigotes. Vaccine 2003, 22, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, C.S.; Giddings, O.K.; Yoshida, N.; Hoft, D.F. Immune responses to gp82 provide protection against mucosal Trypanosoma cruzi infection. Mem. Inst. Oswaldo Cruz 2010, 105, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Serna, C.; Lara, J.A.; Rodrigues, S.P.; Marques, A.F.; Almeida, I.C.; Maldonado, R.A. A synthetic peptide from Trypanosoma cruzi mucin-like associated surface protein as candidate for a vaccine against Chagas disease. Vaccine 2014, 32, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Campos, V.; Martinez-Vega, P.; Ramirez-Sierra, M.J.; Rosado-Vallado, M.; Seid, C.A.; Hudspeth, E.M.; Wei, J.; Liu, Z.; Kwityn, C.; Hammond, M.; et al. Expression, purification, immunogenicity, and protective efficacy of a recombinant Tc24 antigen as a vaccine against Trypanosoma cruzi infection in mice. Vaccine 2015, 33, 4505–4512. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.A.; Versteeg, L.; Wang, Q.; Pollet, J.; Zhan, B.; Gusovsky, F.; Bottazzi, M.E.; Hotez, P.J.; Jones, K.M. A therapeutic vaccine prototype induces protective immunity and reduces cardiac fibrosis in a mouse model of chronic Trypanosoma cruzi infection. PLoS Neglected Trop. Dis. 2019, 13, e0007413. [Google Scholar] [CrossRef] [PubMed]
- Rios, L.E.; Vázquez-Chagoyán, J.C.; Pacheco, A.O.; Zago, M.P.; Garg, N.J. Immunity and vaccine development efforts against Trypanosoma cruzi. Acta Tropica 2019, 200, 105168. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.R.; Almeida, I.C. Carbohydrate immunity in American trypanosomiasis. Springer Semin. Immunopathol. 1993, 15, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Almeida, I.C.; Milani, S.R.; Gorin, P.A.; Travassos, L.R. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-alpha-galactosyl antibodies. J. Immunol. 1991, 146, 2394–2400. [Google Scholar] [CrossRef]
- Almeida, I.C.; Ferguson, M.A.; Schenkman, S.; Travassos, L.R. Lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem. J. 1994, 304, 793–802. [Google Scholar] [CrossRef]
- Almeida, I.C.; Krautz, G.M.; Krettli, A.U.; Travassos, L.R. Glycoconjugates of Trypanosoma cruzi: A 74 kD antigen of trypomastigotes specifically reacts with lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas disease. J. Clin. Lab. Anal. 1993, 7, 307–316. [Google Scholar] [CrossRef]
- Portillo, S.; Zepeda, B.G.; Iniguez, E.; Olivas, J.J.; Karimi, N.H.; Moreira, O.C.; Marques, A.F.; Michael, K.; Maldonado, R.A.; Almeida, I.C. A prophylactic alpha-Gal-based glycovaccine effectively protects against murine acute Chagas disease. NPJ Vaccines 2019, 4, 13. [Google Scholar] [CrossRef]
- Iniguez, E.; Schocker, N.S.; Subramaniam, K.; Portillo, S.; Montoya, A.L.; Al-Salem, W.S.; Torres, C.L.; Rodriguez, F.; Moreira, O.C.; Acosta-Serrano, A.; et al. An alpha-Gal-containing neoglycoprotein-based vaccine partially protects against murine cutaneous leishmaniasis caused by Leishmania major. PLoS Negl. Trop. Dis. 2017, 11, e0006039. [Google Scholar] [CrossRef] [PubMed]
- Parussini, F.; Duschak, V.G.; Cazzulo, J.J. Membrane-bound cysteine proteinase isoforms in different developmental stages of Trypanosoma cruzi. Cell Mol. Biol. 1998, 44, 513–519. [Google Scholar]
- Bhatia, V.; Sinha, M.; Luxon, B.; Garg, N. Utility of the Trypanosoma cruzi Sequence Database for Identification of Potential Vaccine Candidates by In Silico and In Vitro Screening. Infect. Immun. 2004, 72, 6245–6254. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Tarleton Rick, L. Genetic Immunization Elicits Antigen-Specific Protective Immune Responses and Decreases Disease Severity in Trypanosoma cruzi Infection. Infect. Immun. 2002, 70, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Albareda, M.C.; Natale, M.A.; De Rissio, A.M.; Fernandez, M.; Serjan, A.; Alvarez, M.G.; Cooley, G.; Shen, H.; Viotti, R.; Bua, J.; et al. Distinct Treatment Outcomes of Antiparasitic Therapy in Trypanosoma cruzi-Infected Children Is Associated with Early Changes in Cytokines, Chemokines, and T-Cell Phenotypes. Front. Immunol. 2018, 9, 1958. [Google Scholar] [CrossRef]
- Limon-Flores, A.Y.; Cervera-Cetina, R.; Tzec-Arjona, J.L.; Ek-Macias, L.; Sánchez-Burgos, G.; Ramirez-Sierra, M.J.; Cruz-Chan, J.V.; VanWynsberghe, N.R.; Dumonteil, E. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice: Role of CD4+ and CD8+ T cells. Vaccine 2010, 28, 7414–7419. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Scientific Reports 2017, 7, 252. [Google Scholar] [CrossRef]
- Sanchez Alberti, A.; Bivona, A.E.; Matos, M.N.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; González, G.; Morales, C.; Cardoso, A.C.; et al. Mucosal Heterologous Prime/Boost Vaccination Induces Polyfunctional Systemic Immunity, Improving Protection Against Trypanosoma cruzi. Front. Immunol. 2020, 11, 128. [Google Scholar] [CrossRef]
- Araújo, A.F.S.; De Alencar, B.C.G.; Vasconcelos, J.R.C.; Hiyane, M.I.; Marinho, C.R.F.; Penido, M.L.O.; Boscardin, S.B.; Hoft, D.F.; Gazzinelli, R.T.; Rodrigues, M.M. CD8+-T-Cell-Dependent Control of Trypanosoma cruzi Infection in a Highly Susceptible Mouse Strain after Immunization with Recombinant Proteins Based on Amastigote Surface Protein 2. Infect. Immun. 2005, 73, 6017–6025. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Garg, N.J. Prophylactic Efficacy of TcVac2 against Trypanosoma cruzi in Mice. PLoS Neglected Trop. Dis. 2010, 4, e797. [Google Scholar] [CrossRef] [PubMed]
- Rigato, P.O.; De Alencar, B.C.; De Vasconcelos, J.R.C.; Dominguez, M.R.; Araújo, A.F.; Machado, A.V.; Gazzinelli, R.T.; Bruna-Romero, O.; Rodrigues, M.M. Heterologous Plasmid DNA Prime-Recombinant Human Adenovirus 5 Boost Vaccination Generates a Stable Pool of Protective Long-Lived CD8+ T Effector Memory Cells Specific for a Human Parasite, Trypanosoma cruzi. Infect. Immun. 2011, 79, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Mancino, C.; Pollet, J.; Zinger, A.; Jones, K.M.; Villar, M.J.; Leao, A.C.; Adhikari, R.; Versteeg, L.; Tyagi Kundu, R.; Strych, U.; et al. Harnessing RNA Technology to Advance Therapeutic Vaccine Antigens against Chagas Disease. ACS Appl. Mater. Interfaces 2024, 16, 15832–15846. [Google Scholar] [CrossRef] [PubMed]
- Poveda, C.; Leão, A.C.; Mancino, C.; Taraballi, F.; Chen, Y.-L.; Adhikari, R.; Villar, M.J.; Kundu, R.; Nguyen, D.M.; Versteeg, L.; et al. Heterologous mRNA-protein vaccination with Tc24 induces a robust cellular immune response against Trypanosoma cruzi, characterized by an increased level of polyfunctional CD8+ T-cells. Curr. Res. Immunol. 2023, 4, 100066. [Google Scholar] [CrossRef]
- Hunter, D.J.; Abdool Karim, S.S.; Baden, L.R.; Farrar, J.J.; Hamel, M.B.; Longo, D.L.; Morrissey, S.; Rubin, E.J. Addressing Vaccine Inequity—Covid-19 Vaccines as a Global Public Good. N. Engl. J. Med. 2022, 386, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- WHO. Vaccine Inequity Undermining Global Economic Recovery. 2021. Available online: https://www.who.int/news/item/22-07-2021-vaccine-inequity-undermining-global-economic-recovery#:~:text=New%20Global%20Dashboard%20on%20COVID,rate%20as%20high%2Dincome%20countries (accessed on 25 March 2024).
- Jones, K.; Versteeg, L.; Damania, A.; Keegan, B.; Kendricks, A.; Pollet, J.; Cruz-Chan Julio, V.; Gusovsky, F.; Hotez Peter, J.; Bottazzi Maria, E. Vaccine-Linked Chemotherapy Improves Benznidazole Efficacy for Acute Chagas Disease. Infect. Immun. 2018, 86, e00876-17. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Chan, J.V.; Villanueva-Lizama, L.E.; Versteeg, L.; Damania, A.; Villar, M.J.; González-López, C.; Keegan, B.; Pollet, J.; Gusovsky, F.; Hotez, P.J.; et al. Vaccine-linked chemotherapy induces IL-17 production and reduces cardiac pathology during acute Trypanosoma cruzi infection. Sci. Rep. 2021, 11, 3222. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.M.; Mangin, E.N.; Reynolds, C.L.; Villanueva, L.E.; Cruz, J.V.; Versteeg, L.; Keegan, B.; Kendricks, A.; Pollet, J.; Gusovsky, F.; et al. Vaccine-linked chemotherapy improves cardiac structure and function in a mouse model of chronic Chagas disease. Front. Cell Infect. Microbiol. 2023, 13, 1106315. [Google Scholar] [CrossRef]
- Liu, Z.; Ulrich vonBargen, R.; Kendricks, A.L.; Wheeler, K.; Leão, A.C.; Sankaranarayanan, K.; Dean, D.A.; Kane, S.S.; Hossain, E.; Pollet, J.; et al. Localized cardiac small molecule trajectories and persistent chemical sequelae in experimental Chagas disease. Nat. Commun. 2023, 14, 6769. [Google Scholar] [CrossRef]
- Prochetto, E.; Bontempi, I.; Rodeles, L.; Cabrera, G.; Vicco, M.; Cacik, P.; Pacini, M.F.; Pérez Gianeselli, M.; Pérez, A.R.; Marcipar, I. Assessment of a combined treatment with a therapeutic vaccine and benznidazole for the Trypanosoma cruzi chronic infection. Acta Trop. 2022, 229, 106334. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, R.B.; Bocchi, E.A. Shorter treatment in chronic Chagas disease: A new promise? Lancet Infect. Dis. 2024, 24, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.F.; Jayawardhana, S.; Lewis, M.D.; White, K.L.; Shackleford, D.M.; Chen, G.; Saunders, J.; Osuna-Cabello, M.; Read, K.D.; Charman, S.A.; et al. Nitroheterocyclic drugs cure experimental Trypanosoma cruzi infections more effectively in the chronic stage than in the acute stage. Sci. Rep. 2016, 6, 35351. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Liu, X.; Stinson, M.; Rivera, I.; Groessl, T.; Tuntland, T.; Yeh, V.; Wen, B.; Molteni, V.; Glynne, R.; et al. Antitrypanosomal Treatment with Benznidazole Is Superior to Posaconazole Regimens in Mouse Models of Chagas Disease. Antimicrob. Agents Chemother. 2015, 59, 6385–6394. [Google Scholar] [CrossRef]
- Branquinho, R.T.; Mosqueira, V.C.; De Oliveira-Silva, J.C.; Simões-Silva, M.R.; Saúde-Guimarães, D.A.; De Lana, M. Sesquiterpene lactone in nanostructured parenteral dosage form is efficacious in experimental Chagas disease. Antimicrob. Agents Chemother. 2014, 58, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- De Mello, C.G.; Branquinho, R.T.; Oliveira, M.T.; Milagre, M.M.; Saúde-Guimarães, D.A.; Mosqueira, V.C.; Lana, M. Efficacy of Lychnopholide Polymeric Nanocapsules after Oral and Intravenous Administration in Murine Experimental Chagas Disease. Antimicrob. Agents Chemother. 2016, 60, 5215–5222. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Capelari-Oliveira, P.; Bahia, M.T.; Mosqueira, V.C.F. Review on Experimental Treatment Strategies Against Trypanosoma cruzi. J. Exp. Pharmacol. 2021, 13, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.P.; Gazzinelli, R.T.; Morel, C.M.; Precioso, A.R. Why do we still have not a vaccine against Chagas disease? Mem. Inst. Oswaldo Cruz 2022, 117, e200314. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Part 2: Potential Alternatives to the Use of Animals in Preclinical Trials. JACC Basic. Transl. Sci. 2020, 5, 387–397. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C. The Case for the Development of a Chagas Disease Vaccine: Why? How? When? Trop. Med. Infect. Dis. 2021, 6, 16. [Google Scholar] [CrossRef]
- Brown, C.M.; Slavinski, S.; Ettestad, P.; Sidwa, T.J.; Sorhage, F.E. Compendium of Animal Rabies Prevention and Control, 2016. J. Am. Vet. Med. Assoc. 2016, 248, 505–517. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Name | Treatment Regimen | Stage of Disease | Target Patients | Country | Status |
|---|---|---|---|---|---|
| CHICO SECURE (Chagas disease in children treated with Nifurtimox with follow-up for seroconversion and cure) [20] | NFX 8–20 mg/kg/day (30 days) NFX 8–20 mg/kg/day (60 days) | Acute and chronic | Pediatric patients <18 years old | Multicentric (Argentina, Bolivia and Colombia) | Ongoing |
| TESEO (new therapies and biomarkers for Chagas disease) [21] | BZN 300 mg/day (60 days) BZN 150 mg/day (30 days) BZN 150 mg/day (90 days) NFX 480 mg/day (60 days) NFX 480 mg/day (30 days) NFX 240 mg/day (90 days) | Indeterminate or early cardiac compromised | Adult patients (18–50 years old) | Bolivia | Ongoing |
| BETTY (Short-course Benznidazole treatment to reduce Trypanosoma cruzi parasitic load in women of reproductive age) [22] | BZN 300 mg/day (60 days) BZN 150 mg/day (30 days) | Chronic in postpartum period | Postpartum T. cruzi positive women patients | Argentina | Ongoing |
| EQUITY [23] | BZN 300 mg/day (60 days) BZN 150 mg/day (120 days) NFX 480 mg/day (60 days) NFX 240 mg/day (120 days) | Chronic Indeterminate | Adult patients (20–65 years old) | Colombia | Ongoing |
| MULTIBENZ (Evaluation of different Benznidazole regimens for the treatment of chronic Chagas disease) [24,25] | BZN 300 mg/day (60 days) BZN 400 mg/day (15 days) BZN 150 mg/day (60 days) | Chronic | Adult patients (>18 years old) | Multicentric (Spain, Brazil, Argentina, Colombia) | Completed |
| BENEFIT (Benznidazole evaluation for interrupting trypanosomiasis) [26] | BZN 300 mg/day (80 days) | Chronic cardiomyopathy | Adult patients (18–75 years old) | Multicentric (Brazil, Argentina, Colombia, Bolivia and El Salvador) | Completed |
| TRAENA (Tratamiento con benznidazol en pacientes adultos con enfermedad de Chagas crónica de bajo riesgo) [27] | BZN 200 mg/day (60 days) | Chronic | Adult patients (20–55 years old) | Argentina | Completed |
| Name | Medication and Regimen | Target Patients | Target Patients | Country | Status |
|---|---|---|---|---|---|
| BENDITA (Benznidazole; new doses, improved treatment, and therapeutic associations) [33] | BZN 300 mg/day (60 days) BZN 300 mg/day (30 days) BZN 300 mg/day (15 days) BZN 150 mg/day (30 days) BZN 150 mg/day (30 days) + Forsvuconazole (300 mg/day 3 days + 300 mg/week) BZN 150 mg/week (60 days) + Forsvuconazole (300 mg/day 3 days + 300 mg/week) | Chronic Indeterminate | Adult patients (18–50 years old) | Bolivia | Completed |
| E1224 [34] | E1224 4000 mg (8 weeks) E1224 2000 mg (8 weeks) E1224 2400 mg (4 weeks) + Placebo (4 weeks) BZN 5 mg/kg/day (60 days) Placebo (8 weeks) | Chronic Indeterminate | Adult patients (18–50 years old) | Bolivia | Completed |
| STOP-CHAGAS [35] | Posoconazole 800 mg/day (60 days) BZN 400 mg/day + Placebo 10 mg/day (60 days) Posoconazole 800 mg/day + BZN 400 mg/day (60 days) Placebo 10 mg/day (60 days) | Chronic Indeterminate | Adult patients (18–50 years old) | Multicentric (Argentina, Chile, Colombia Guatemala, Mexico, and Spain) | Completed |
| CHAGASAZOL [36] | Posaconazole 800 mg/day (60 days) Posaconazole 200 mg/day (60 days) BZN 300 mg/day (60 days) | Chronic Indeterminate | Adult patients (>18 years old) | Multicentric (Bolivia, Brazil and Paraguay) | Completed |
| Oral Fexinidazole Dosing Regimens for the Treatment of Adults With Chronic Indeterminate Chagas disease [31,37] | Fexinidazole 600 mg/day (10 days) Fexinidazole 1200 mg/day (3 days) + Placebo 1200 mg/day (7 days) Fexinidazole 600 mg/day (3 days) + Fexinidazole 1200 mg/day (4 days) + Placebo (3 days) | Chronic Indeterminate | Adult patients (18–60 years old) | Spain | Completed |
| Vaccine Antigen | Mouse Model/Infection Route/T. cruzi Strain | Dose/ Immunization Route | Parasite Burden /Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|
| Cruzipain [54] | BALBc/ SC/Tulahuen | 2 doses, 1 week apart/IM | Reduction/ Increase | Cruzipain-specific IgG (Sera) and secretory IgA (Fecal) | Cytolytic activity Increased CD8+ and levels of IFN-γ |
| ASP-2 or UB-ASP-2 [55] | C57BL/6-PA28 knockout (PA28α/β−/−) and LMP2 or LMP7 knockout (LMP2−/− or LMP7−/−)/SC/Tulahuen | 4 doses, 2-week intervals /SC | Reduction/ Increase | NR | Cytolytic activity Increased levels CD8+ and IFN-γ |
| TcSSP4 [56] | BALBc/IP/H8 | 2 doses, 2-week intervals/IP | Reduction/ Increase | NR | Increased levels IL-10 and IFN-γ |
| Transialidase [57,58] | BALBc/ SC/Tulahuen | 2 doses, 1 week apart/IN | Increase | TS-specific IgG (Sera) and secretory IgA (Fecal) | Increased levels CD8+ and IFN-γ |
| TS + IL-15 [59] | BALBc/SC/Tulahuen | 3 doses, 2-weeks intervals/IM | NR/Increase | No differences | Increased levels CD8+ and IFN-γ |
| Vaccine Antigen | Mouse Model/Infection Route/T. cruzi Strain | Dose/ Immunization Route | Parasite Burden/Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|
| Adenovirus expressing TSSA CD8+ epitope [60] | C57BL/6/IP/Tulahuen lethal and sublethal | 2 doses 12 days apart/Prime IM and boost IM or IP | Reduction/ Increase | NR | Increased levels IFN-γ |
| Adenovirus expressing TS and ASP-2 [61] | BALBc and C57BL/6/ IP/Y | Multivalent 2 doses, 6- to 8-week interval/SC | Reduction/ Increase | NR | Cytolytic activity Increased levels IFN-γ |
| Vaccine Antigen | Mouse model/Infection Route/T. cruzi Strain | Scheme/Dose/ Immunization Route | Parasite Burden/Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|
| ASP-2 [64] | A/Sn, C3H/HeJ and C3H/HePAS/ IP/Y | Prophylactic/3 doses at 0, 2 and 4 weeks/IP | Reduction/ Increase | NR | Increased CD8+ |
| rCruzipain Adjuvant: CpG ODN [65,66] | C3H/HeN (H-2K haplotype)/ NR/RA | Prophylactic/2 doses, 1 week apart/IM | Reduction/ NR | IgG | Increased levels IFN-γ, IL-2, and IL-10 |
| rGP82 Adjuvant: CpG ODN [67] | BALBc/ conjunctival or oral/ Tulahuen | Prophylactic/2 doses, 2 weeks apart/IN | Reduction/ Increase | NR | Increased levels IFN-γ |
| MASPpep-KLH Adjuvant: Al(OH)3 [68] | C3H/HeNsd; BALBc/ IP/Y | Prophylactic/3 doses, 10–15 days/IP | Reduction/ Increase | IgG | Increased levels IL-4, IL-10, IFN-γ and IL-12 |
| rTc24 Adjuvant: E6020-SE/MPLA/CpG [58,69,70] | BALBc/IP/H1 | Prophylactic/2 doses, 2 weeks apart/IM | Reduction/ Increase | IgG2a | Increased levels IFN-γ |
| BALBc/IP/H1 | Therapeutic/2 doses, 4 weeks apart/SC | Reduction/ NR | NR | Increased CD8+, Increased levels IFN-γ | |
| ICR/IP/H1 | Therapeutic/2 doses, 2 weeks apart/SC | Reduction/ NR | Balanced IgG1/IgG2a | Increased levels IFN-γ | |
| TS Adjuvant: CpG ODN [41,71] | BALBc/ SC/Tulahuen | Prophylactic/2 doses, 1 week apart/IN | Reduction/ Increase | TS-specific IgG (Sera) and secretory IgA (Fecal) | Increased CD8+ and levels of IFN-γ |
| Vaccine Antigen | Mouse Model/Infection Route/T. cruzi Strain | Scheme/Dose/ Immunization Route | Parasite Burden/Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|
| Galα3LN-HSA [76] | C57BL/6/IP/CL Brener | Prophylactic/4 doses at 0, 1, 2 and 3 weeks/IP | Reduction/ Increase | IgG, IgG1, IgG2b, IgG3 | Increased CD4+, Increased CD8+, Increased CD4+ CD44+ |
| Vaccine Antigen | Vaccine Platform | Mouse Model/ Infection Route/T. cruzi Strain | Scheme/Dose/ Immunization Route | Parasite Burden/Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|---|
| TS and ASP-2 [61] | Viral vector | BALBc and C57BL/6/ NR/Y | Prophylactic/SC 2 doses, 6- to 8-week interval/SC | Reduction/ Increase | NR | Cytolytic activity Increased levels IFN-γ |
| TSA-1, ASP-1 and ASP-2 with IL-12 and GM-CS [80] | DNA | C57BL/6/IP/Brazil | Prophylactic/ 2 doses, 6 weeks apart/IM | Reduction/ Increase | Antibodies | Cytolytic activity |
| TSA-1 and Tc24 [82] | DNA | BALBc/IP/H1 | Prophylactic and therapeutic/2 doses, 2 weeks apart/NR | Reduction | NR | Increase antigen specific T cells. |
| BALBc and C57BL/6/IP/ H1 | Therapeutic/2 doses, 2 weeks apart during acute phase/NR | Reduction | NR | NR | ||
| ICR/IP/H1 | Therapeutic/2 doses, 2 weeks apart during chronic phase/NR | NR | NR | Increase CD8+ |
| Vaccine Antigen | Vaccine Platform | Animal Model/ Infection Route/T. cruzi Strain | Scheme/Dose/Immunization Route | Parasite Burden/Survival | Antibody Response | Cellular Immune Response |
|---|---|---|---|---|---|---|
| rASP2 [85] | Recombinant Protein DNA | A/Sn inbreed/ IP/Y | Prophylactic Protein/DNA/3 doses at 0, 3 and 5 weeks/IP | Reduction/ Increase | NR | Increased CD8+, IFN-γ |
| TcVac2 (TcG1, TcG2, TcG4) + IL-12 + GM-CSF [86] | DNA and recombinant protein boost + saponin | C57BL/6/IP/ Sylvio X10/4 | Prophylactic/5 doses, 3 of DNA vaccine with 2 weeks interval and 2 doses of recombinant protein/IM | Reduction/ NR | Increased IgG, IgG1, and IgG2b | Increased CD8+, IFN-γ |
| DNA + adenovirus expressing TS and ASP-2 clone 9 (Prime-Boost) [87] | DNA and viral vector | C57BL/6 and A/Sn/Y/SC | Prophylactic/DNA Prime and viral vector 21 days after prime vaccination/IM | Reduction/ Increase | NR | Cytolytic activity Increased levels CD8+ and IFN-γ |
| Traspain + CDA, Nt-Cz + ASP2 + CDA [84] | DNA and protein | C3H/HeN (H-2k)/Lethal RA and chronic Clone K-68/IP | Prophylactic/4 doses, 2 doses of each antigen, dose interval of 10 days/Prime PO − boost IN | Reduction/ Increase | NR | Increased polyfunctional cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farani, P.S.G.; Jones, K.M.; Poveda, C. Treatments and the Perspectives of Developing a Vaccine for Chagas Disease. Vaccines 2024, 12, 870. https://doi.org/10.3390/vaccines12080870
Farani PSG, Jones KM, Poveda C. Treatments and the Perspectives of Developing a Vaccine for Chagas Disease. Vaccines. 2024; 12(8):870. https://doi.org/10.3390/vaccines12080870
Chicago/Turabian StyleFarani, Priscila Silva Grijó, Kathryn Marie Jones, and Cristina Poveda. 2024. "Treatments and the Perspectives of Developing a Vaccine for Chagas Disease" Vaccines 12, no. 8: 870. https://doi.org/10.3390/vaccines12080870