

SARS-CoV-2 Vaccination Responses in Anti-CD20-Treated Progressive Multiple Sclerosis Patients Show Immunosenescence in Antigen-Specific B and T Cells
 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Methods
2.1. Patient Selection
2.2. Blood Collection and Isolation of Mononuclear Cells
2.3. Activation Induced Cell Marker Assay (AIM) and T Cell Phenotype
2.4. Detection of SARS-CoV-2-Specific B Cells
2.5. Intracellular Cytokine Staining (ICS)
2.6. Computational Analysis of Flow Cytometry Data
2.6.1. T Cell Analysis
2.6.2. B Cell Analysis
2.7. Statistical Analysis
2.8. Principal Component Analysis
3. Results
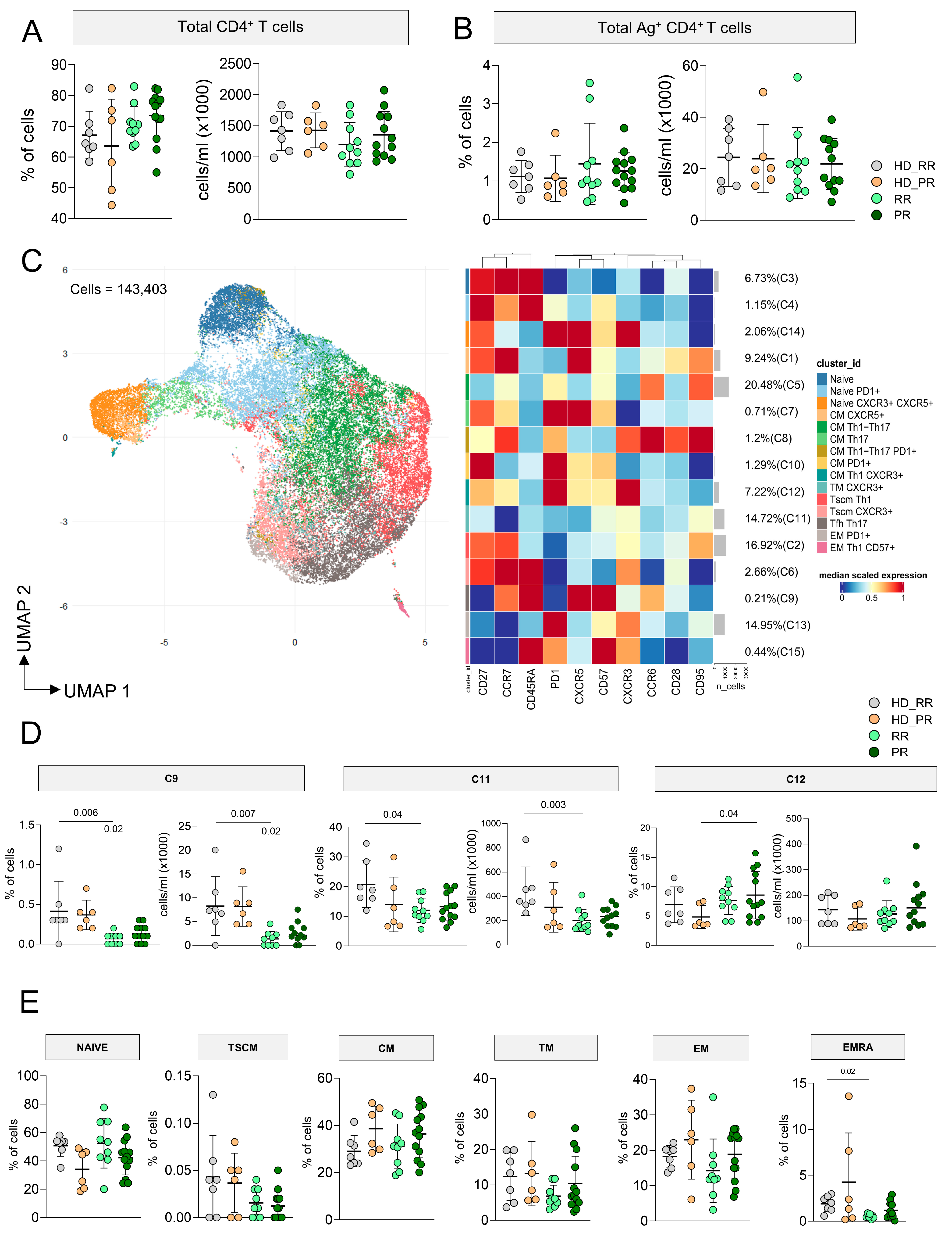
3.1. MS Patients Undergoing aCD20 Therapy Develop a CD4+ Ag+ Specific T Cell Response Similar to Healthy Donors
3.2. Progressive MS Patients Display High Percentage of CD4+CD107a+ Ag+ Specific T Cell
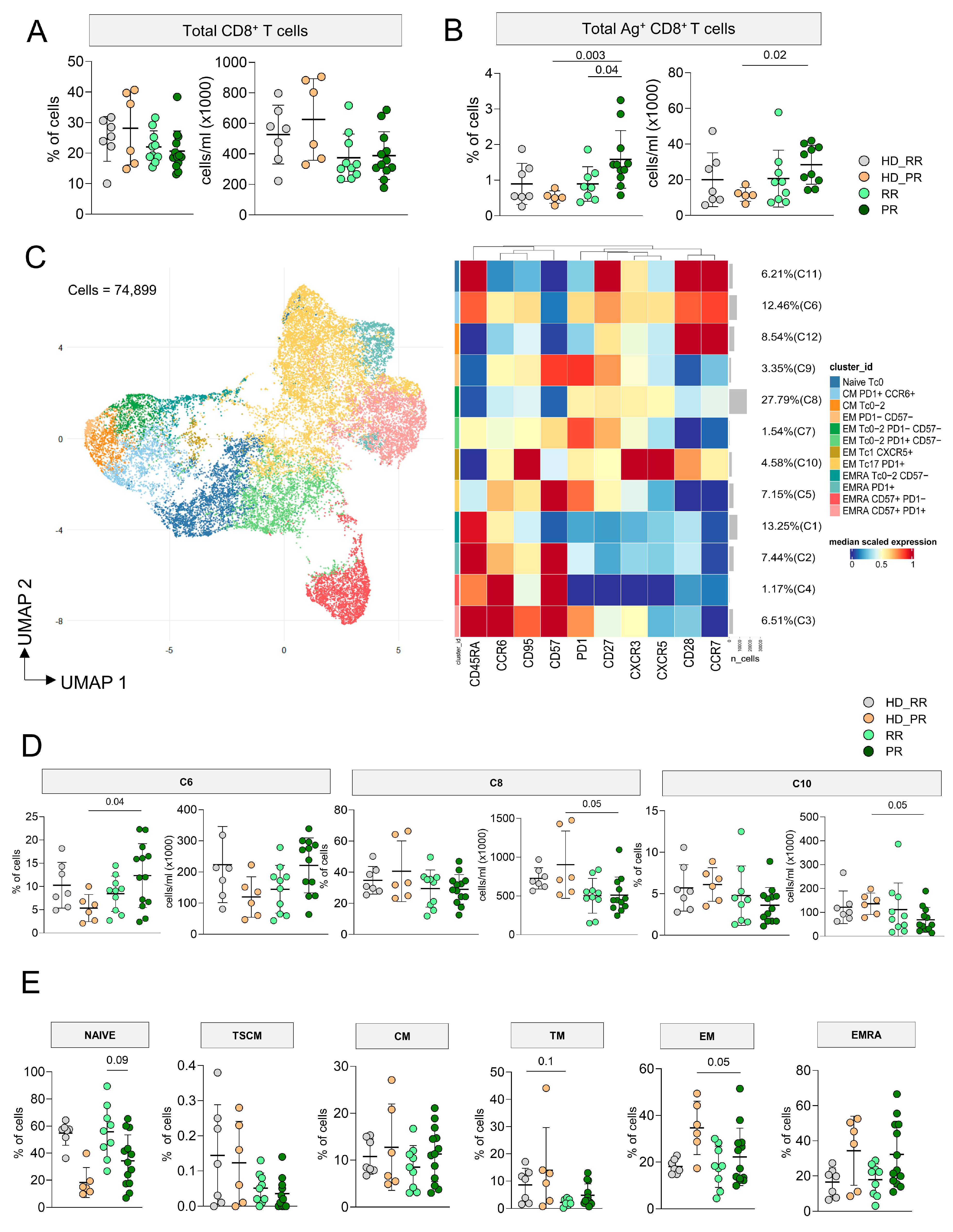
3.3. Higher Percentage of CD8+ Ag+ T Cells Characterize Progressive Patients
3.4. CD8+ Ag+ T Cells from PR Patients Are Much More Polyfunctional If Compared to Age-Matched HD
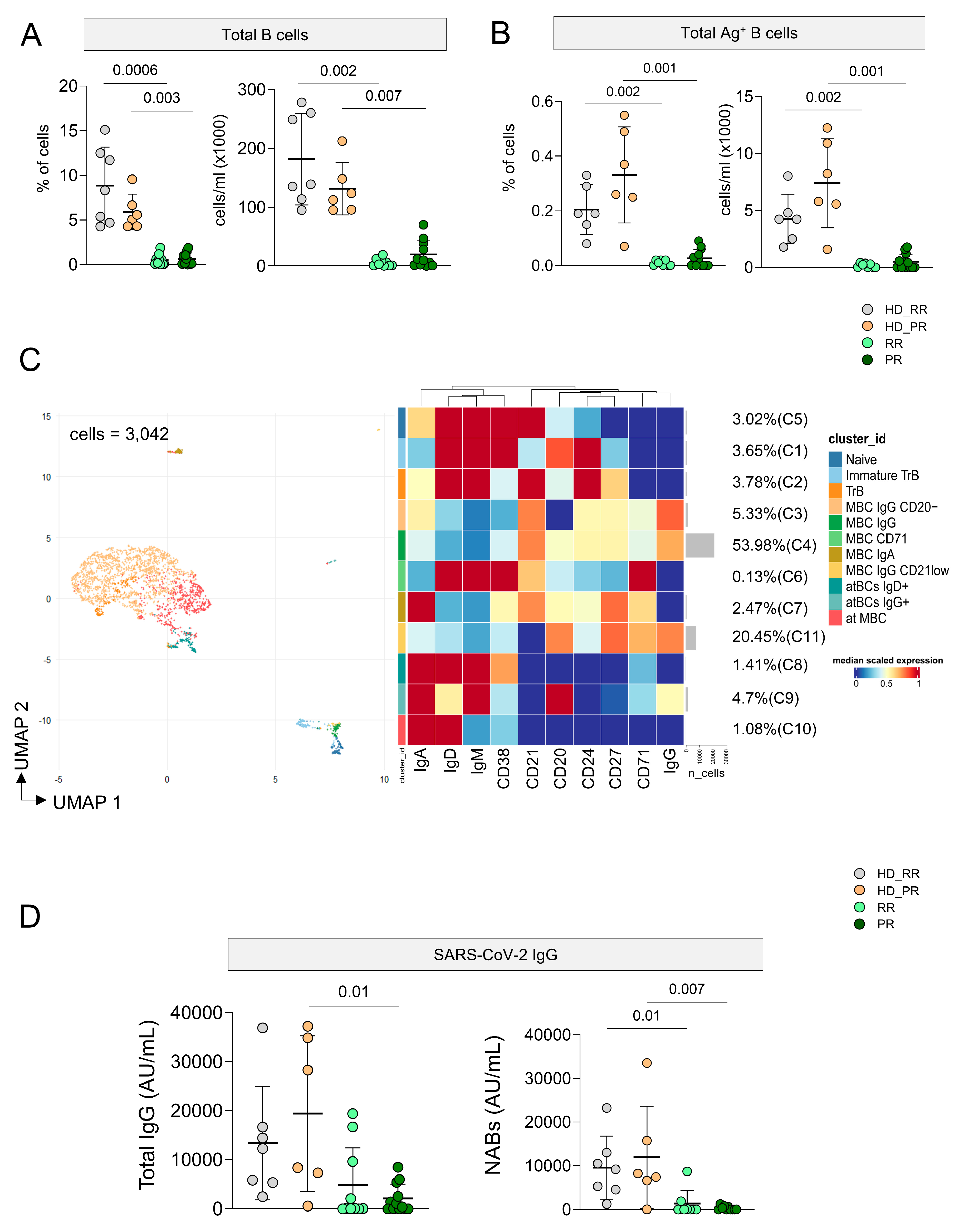
3.5. Ag+ B Cell Response Is Low in MS Patients Undergoing aCD20 Treatment, but It Is Still Detectable
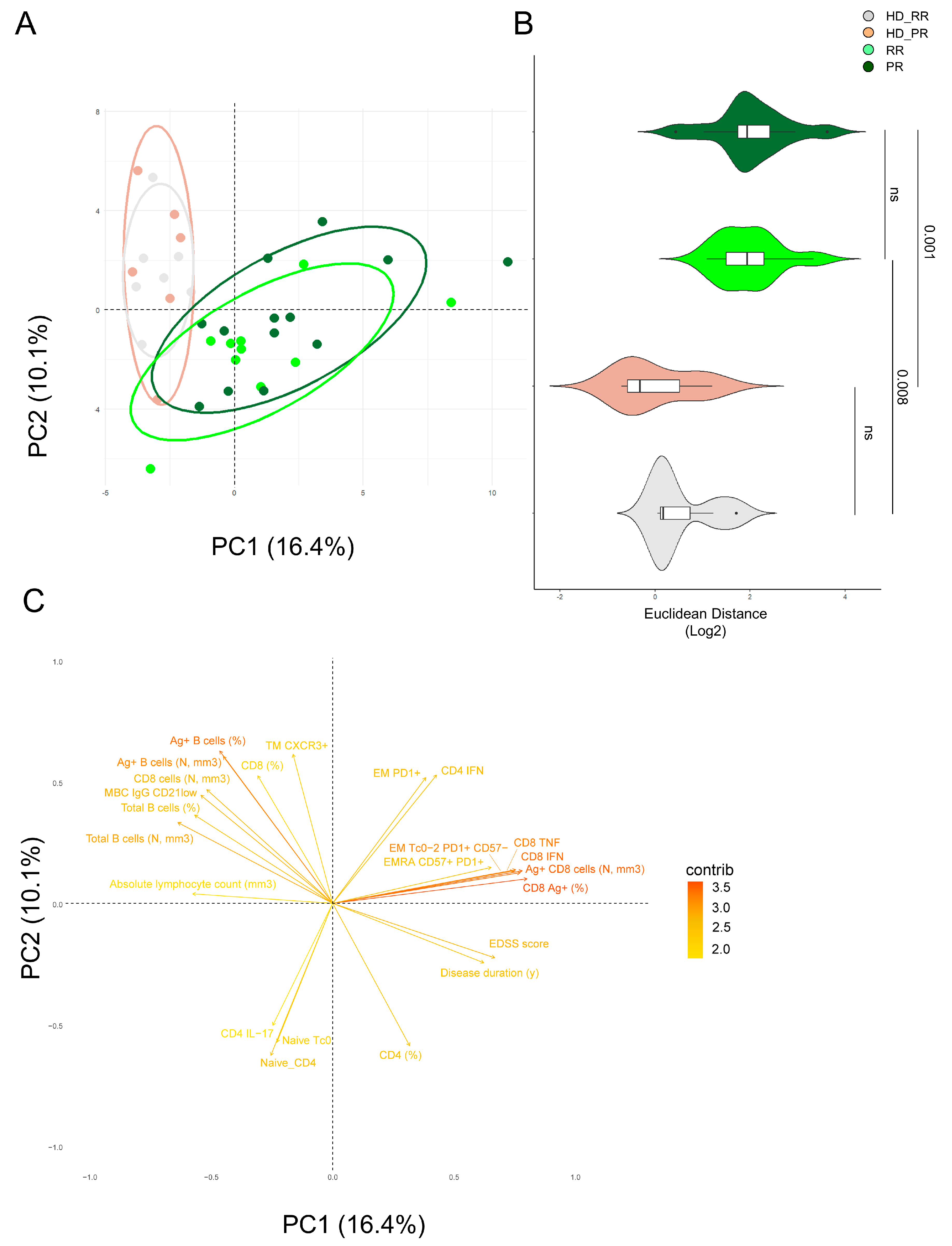
3.6. Principal Component Analysis Revealed That Relapsing Remitting and Progressive MS Patients Treated with Anti-CD20 Showed Similar SARS-CoV-2 Immune Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Filippi, M.P.P.; Barkhof, F.; Ciccarelli, O.; Cossarizza, A.; De Stefano, N.; Gasperini, C.; Geraldes, R.; Granziera, C.; Haider, L.; Lassmann, H.; et al. The ageing central nervous system in multiple sclerosis: The imaging perspective. Brain 2024, awae251, Online ahead of print. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, O.; Barkhof, F.; Bodini, B.; De Stefano, N.; Golay, X.; Nicolay, K.; Pelletier, D.; Pouwels, P.J.; Smith, S.A.; Wheeler-Kingshott, C.A.; et al. Pathogenesis of multiple sclerosis: Insights from molecular and metabolic imaging. Lancet Neurol. 2014, 13, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.-P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Bar-Or, A. Emerging therapies to target CNS pathophysiology in multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Preziosa, P.; Scalfari, A.; Colato, E.; Marastoni, D.; Absinta, M.; Battaglini, M.; De Stefano, N.; Di Filippo, M.; Hametner, S.; et al. Determinants and Biomarkers of Progression Independent of Relapses in Multiple Sclerosis. Ann. Neurol. 2024, 96, 1–20. [Google Scholar] [CrossRef]
- Woo, M.S.; Engler, J.B.; Friese, M.A. The neuropathobiology of multiple sclerosis. Nat. Rev. Neurosci. 2024, 25, 493–513. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis—From targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Margoni, M.; Preziosa, P.; Filippi, M.; Rocca, M.A. Anti-CD20 therapies for multiple sclerosis: Current status and future perspectives. J. Neurol. 2022, 269, 1316–1334. [Google Scholar] [CrossRef] [PubMed]
- de Sèze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef] [PubMed]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Martinez-Caceres, E.M.; Rio, J.; Barbera, N.; Marzo, M.E.; Montalban, X. Immunological profile of patients with primary progressive multiple sclerosis. Expression of adhesion molecules. Brain 1999, 122 Pt 12, 2297–2307. [Google Scholar] [CrossRef]
- De Biasi, S.; Simone, A.M.; Bianchini, E.; Lo Tartaro, D.; Pecorini, S.; Nasi, M.; Patergnani, S.; Carnevale, G.; Gibellini, L.; Ferraro, D.; et al. Mitochondrial functionality and metabolism in T cells from progressive multiple sclerosis patients. Eur. J. Immunol. 2019, 49, 2204–2221. [Google Scholar] [CrossRef]
- De Biasi, S.; Lo Tartaro, D.; Neroni, A.; Rau, M.; Paschalidis, N.; Borella, R.; Santacroce, E.; Paolini, A.; Gibellini, L.; Ciobanu, A.L.; et al. Immunosenescence and vaccine efficacy revealed by immunometabolic analysis of SARS-CoV-2-specific cells in multiple sclerosis patients. Nat. Commun. 2024, 15, 2752. [Google Scholar] [CrossRef]
- Guerrera, G.; Mandelli, A.; Finardi, A.; Orrico, M.; D’Orso, S.; Picozza, M.; Noviello, M.; Beretta, V.; Bonetti, B.; Calabrese, M.; et al. Anti-SARS-CoV-2 T-stem cell memory persists in ocrelizumab-treated MS patients. Mult. Scler. 2022, 28, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Achiron, A.; Mandel, M.; Dreyer-Alster, S.; Harari, G.; Magalashvili, D.; Sonis, P.; Dolev, M.; Menascu, S.; Flechter, S.; Falb, R.; et al. Author response to: Correspondence to humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211020082. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Reyes-Mantilla, M.; Jank, L.; Harris, S.; Douglas, M.; Smith, M.D.; Calabresi, P.A.; Mowry, E.M.; Fitzgerald, K.C.; Bhargava, P. Discordant humoral and T cell immune responses to SARS-CoV-2 vaccination in people with multiple sclerosis on anti-CD20 therapy. EBioMedicine 2021, 73, 103636. [Google Scholar] [CrossRef]
- Gallo, A.; Capuano, R.; Donnarumma, G.; Bisecco, A.; Grimaldi, E.; Conte, M.; D’Ambrosio, A.; Galdiero, M.; Tedeschi, G. Preliminary evidence of blunted humoral response to SARS-CoV-2 mRNA vaccine in multiple sclerosis patients treated with ocrelizumab. Neurol. Sci. 2021, 42, 3523–3526. [Google Scholar] [CrossRef]
- Disanto, G.; Galante, A.; Cantu, M.; Sacco, R.; Mele, F.; Eisler, J.J.; Keller, F.; Bernasconi, E.; Sallusto, F.; Zecca, C.; et al. Longitudinal Postvaccine SARS-CoV-2 Immunoglobulin G Titers, Memory B-Cell Responses, and Risk of COVID-19 in Multiple Sclerosis Over 1 Year. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200043. [Google Scholar] [CrossRef]
- Apostolidis, S.A.; Kakara, M.; Painter, M.M.; Goel, R.R.; Mathew, D.; Lenzi, K.; Rezk, A.; Patterson, K.R.; Espinoza, D.A.; Kadri, J.C.; et al. Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat. Med. 2021, 27, 1990–2001. [Google Scholar] [CrossRef]
- Jeantin, L.; Abdi, B.; Soulie, C.; Sterlin, D.; Maillart, E.; Beigneux, Y.; Hippolyte, A.; Belin, L.; Marcelin, A.-G.; Pourcher, V.; et al. Is vaccine response to SARS-CoV-2 preserved after switching to anti-CD20 therapies in patients with multiple sclerosis or related disorders? J. Neurol. Neurosurg. Psychiatry 2023, 95, 19–28. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.; Radbruch, A.; Abrignani, S.; Addo, R.; Akdis, M.; Andrä, I.; Andreata, F.; Annunziato, F.; Arranz, E.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 2021, 51, 2708–3145. [Google Scholar] [CrossRef] [PubMed]
- Paolini, A.; Borella, R.; Neroni, A.; Tartaro, D.L.; Mattioli, M.; Fidanza, L.; Di Nella, A.; Santacroce, E.; Gozzi, L.; Busani, S.; et al. Patients Recovering from Severe COVID-19 Develop a Polyfunctional Antigen-Specific CD4+ T Cell Response. Int. J. Mol. Sci. 2022, 23, 8004. [Google Scholar] [CrossRef]
- Lo Tartaro, D.; Paolini, A.; Mattioli, M.; Swatler, J.; Neroni, A.; Borella, R.; Santacroce, E.; Di Nella, A.; Gozzi, L.; Busani, S.; et al. Detailed characterization of SARS-CoV-2-specific T and B cells after infection or heterologous vaccination. Front. Immunol. 2023, 14, 1123724. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Paolini, A.; Lo Tartaro, D.; Gibellini, L.; Cossarizza, A. Analysis of Antigen-Specific T and B Cells for Monitoring Immune Protection Against SARS-CoV-2. Curr. Protoc. 2023, 3, e636. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Mattioli, M.; Meschiari, M.; Tartaro, D.L.; Paolini, A.; Borella, R.; Neroni, A.; Fidanza, L.; Busani, S.; Girardis, M.; et al. Prognostic immune markers identifying patients with severe COVID-19 who respond to tocilizumab. Front. Immunol. 2023, 14, 1123807. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Wen, B.; Xie, T.; Tang, P.; Hu, Y.; Huang, L.; Jin, K.; Zhang, P.; Liu, Z.; et al. Circulating CXCR3(+) Tfh cells positively correlate with neutralizing antibody responses in HCV-infected patients. Sci. Rep. 2019, 9, 10090. [Google Scholar] [CrossRef]
- Brand, J.S.; Smith, K.A.; Piehl, F.; Olsson, T.; Montgomery, S. Risk of serious infections in multiple sclerosis patients by disease course and disability status: Results from a Swedish register-based study. Brain Behav. Immun. Health 2022, 22, 100470. [Google Scholar] [CrossRef] [PubMed]
- Januel, E.; Hajage, D.; Labauge, P.; Maillart, E.; De Sèze, J.; Zephir, H.; Pelletier, J.; Guilloton, L.; Bensa, C.; Heinzlef, O.; et al. Association Between Anti-CD20 Therapies and COVID-19 Severity Among Patients With Relapsing-Remitting and Progressive Multiple Sclerosis. JAMA Netw. Open 2023, 6, e2319766. [Google Scholar] [CrossRef]
- Vaughn, C.B.; Jakimovski, D.; Kavak, K.S.; Ramanathan, M.; Benedict, R.H.B.; Zivadinov, R.; Weinstock-Guttman, B. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat. Rev. Neurol. 2019, 15, 329–342. [Google Scholar] [CrossRef]
- Amoriello, R.; Mariottini, A.; Ballerini, C. Immunosenescence and Autoimmunity: Exploiting the T-Cell Receptor Repertoire to Investigate the Impact of Aging on Multiple Sclerosis. Front. Immunol. 2021, 12, 799380. [Google Scholar] [CrossRef] [PubMed]
- Dema, M.; Eixarch, H.; Villar, L.M.; Montalban, X.; Espejo, C. Immunosenescence in multiple sclerosis: The identification of new therapeutic targets. Autoimmun. Rev. 2021, 20, 102893. [Google Scholar] [CrossRef]
- Perdaens, O.; van Pesch, V. Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopathogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 2022, 12, 811518. [Google Scholar] [CrossRef]
- Chen, J.; Deng, J.C.; Goldstein, D.R. How aging impacts vaccine efficacy: Known molecular and cellular mechanisms and future directions. Trends Mol. Med. 2022, 28, 1100–1111. [Google Scholar] [CrossRef]
- Lee, J.L.; Linterman, M.A. Mechanisms underpinning poor antibody responses to vaccines in ageing. Immunol. Lett. 2022, 241, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Rezk, A.; Shinoda, K.; Espinoza, D.A.; Elyahu, Y.; Zhang, B.; Chen, A.A.; Shinohara, R.T.; Jacobs, D.; Alcalay, R.N.; et al. Immune aging in multiple sclerosis is characterized by abnormal CD4 T cell activation and increased frequencies of cytotoxic CD4 T cells with advancing age. EBioMedicine 2022, 82, 104179. [Google Scholar] [CrossRef] [PubMed]
- Thakolwiboon, S.; Mills, E.A.; Yang, J.; Doty, J.; Belkin, M.I.; Cho, T.; Schultz, C.; Mao-Draayer, Y. Immunosenescence and multiple sclerosis: Inflammaging for prognosis and therapeutic consideration. Front. Aging 2023, 4, 1234572. [Google Scholar] [CrossRef] [PubMed]
- Goyne, C.E.; Fair, A.E.; Sumowski, P.E.; Graves, J.S. The Impact of Aging on Multiple Sclerosis. Curr. Neurol. Neurosci. Rep. 2024, 24, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Elyahu, Y.; Feygin, I.; Pinkas, N.; Zemer, A.; Shicht, A.; Berner, O.; Eremenko, E.; Nemirovsky, A.; Reshef, K.; Roitman, L.; et al. CD4 T Cells Acquire Cytotoxic Properties to Modulate Cellular Senescence and Aging. bioRxiv 2024. [Google Scholar] [CrossRef]
- Devarajan, P.; Vong, A.M.; Castonguay, C.H.; Silverstein, N.J.; Kugler-Umana, O.; Bautista, B.L.; Kelly, K.A.; Luban, J.; Swain, S.L. Cytotoxic CD4 development requires CD4 effectors to concurrently recognize local antigen and encounter type I IFN-induced IL-15. Cell Rep. 2023, 42, 113182. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Kuo, H.-H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Kouno, T.; Ikawa, T.; Hayatsu, N.; Miyajima, Y.; Yabukami, H.; Terooatea, T.; Sasaki, T.; Suzuki, T.; Valentine, M.; et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl. Acad. Sci. USA 2019, 116, 24242–24251. [Google Scholar] [CrossRef] [PubMed]
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNAvaccine. Nature 2021, 597, 268–273. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Methot, N.; Yu, D.E.; Zhang, Y.; Dan, J.M.; Goodwin, B.; Rubiro, P.; Sutherland, A.; Wang, E.; et al. Impact of SARS-CoV-2 variants on the total CD4+ and CD8+ T cell reactivity in infected or vaccinated individuals. Cell Rep. Med. 2021, 2, 100355. [Google Scholar] [CrossRef]
- Radomir, L.; Kramer, M.P.; Perpinial, M.; Schottlender, N.; Rabani, S.; David, K.; Wiener, A.; Lewinsky, H.; Becker-Herman, S.; Aharoni, R.; et al. The survival and function of IL-10-producing regulatory B cells are negatively controlled by SLAMF5. Nat. Commun. 2021, 12, 1893. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Stolp, J.; Juvet, S.C.; Beckett, J.; Macklin, P.S.; Issa, F.; Hester, J.; Wood, K.J. Ex vivo-expanded human CD19+TIM-1+ regulatory B cells suppress immune responses in vivo and are dependent upon the TIM-1/STAT3 axis. Nat. Commun. 2022, 13, 3121. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, Q.; Gu, X.; Ren, L.; Huang, T.; Li, Y.; Zhang, H.; Liu, Y.; Zhong, J.; Wang, X.; et al. Durability and cross-reactive immune memory to SARS-CoV-2 in individuals 2 years after recovery from COVID-19: A longitudinal cohort study. Lancet Microbe 2024, 5, e24–e33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Population (N = 36) | HD_RR (N = 7) | HD_PR (N = 6) | RR (N = 10) | PR (N = 13) |
|---|---|---|---|---|
| Age, median (IQR), y | 35.0 (29.0–42.5) | 57.5 (55.5–66.25) | 35.5 (30.25–38.0) | 57.0 (53.0–61.0) |
| Females, n (%) | 6.0 (85.7) | 4.0 (66.7) | 6.0 (60.0) | 10.0 (76.9) |
| Disease duration, median (IQR), y | ─ | ─ | 11.5 (10.0–12.75) | 17.0 (10.0–24.00) |
| Disability by EDSS score, median (IQR) | ─ | ─ | 2.75 (1.25–4.38) | 6.0 (4.00–6.50) |
| Time from last treatment starts, median (IQR), y | ─ | ─ | 0.526 (0.50–0.57) | 0.528 (0.50–0.60) |
| Time from last infusion to last vaccination, median (IQR), months | ─ | ─ | 3.70 (0.19–4.81) | 3.41 (1.44–4.33) |
| Absolute lymphocyte count, median (IQR), 103cells/mm3 | 2.22 (1.95–2.33) | 2.225 (2.23–2.23) | 1.61 (1.38–2.06) | 1.74 (1.49–2.22) |
| CD19 B─cell count, median (IQR), n/mm3 | 138,944 (124,511–254,738) | 118,148 (99,847–142,122) | 4381 (1010–11,212) | 7822 (1125–30,770) |
| Breakthrough COVID─19 after full vaccination, n (%) | 2.00 (66.70) | NA | 1.00 (10.00) | 2.00 (15.40) |
| SARS─CoV─2 IgGII titer after full vaccination, AU/mL | ||||
| Median (IQR) | 12,288.50 (5334–16,687.50) | 28,306.60 (7348.60–34,873.30) | 135.65 (41.80–9656) | 1089.1 (9.25–7176.0) |
| SARS─CoV─2 RBD IgG titer after full vaccination, AU/mL | ||||
| Median (IQR) | 9167.221 (4539.23–13,011.16) | 7829.5923 (6621.08–15,762.73) | 83.02 (4.80–8700.06) | 346.13 (0.41–3760.41) |
| Time from first vaccination to sampling, median (IQR), months | 14.25 (9.10–16.56) | 10.71 (7.26–12.83) | 11.67 (9.52–15.40) | 10.94 (9.63–15.01) |
| Time from last vaccination to sampling, median (IQR), months | 3.61 (1.74–6.60) | 3.28 (0.92–6.04) | 2.56 (1.695–8.22) | 2.96 (2.20–7.705) |
| Type of third vaccine dose | ||||
| BNT162b2, n (%) | 5 (71.43%) | 3 (50.00%) | 8 (80.00%) | 9 (69.23%) |
| mRNA─1273, n (%) | 2 (28.57%) | 3 (50.00%) | 2 (20.00%) | 4 (30.77%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Biasi, S.; Ciobanu, A.L.; Santacroce, E.; Lo Tartaro, D.; Degliesposti, G.; D’Angerio, M.; Leccese, M.; Cardi, M.; Trenti, T.; Cuccorese, M.; et al. SARS-CoV-2 Vaccination Responses in Anti-CD20-Treated Progressive Multiple Sclerosis Patients Show Immunosenescence in Antigen-Specific B and T Cells. Vaccines 2024, 12, 924. https://doi.org/10.3390/vaccines12080924
De Biasi S, Ciobanu AL, Santacroce E, Lo Tartaro D, Degliesposti G, D’Angerio M, Leccese M, Cardi M, Trenti T, Cuccorese M, et al. SARS-CoV-2 Vaccination Responses in Anti-CD20-Treated Progressive Multiple Sclerosis Patients Show Immunosenescence in Antigen-Specific B and T Cells. Vaccines. 2024; 12(8):924. https://doi.org/10.3390/vaccines12080924
Chicago/Turabian StyleDe Biasi, Sara, Alin Liviu Ciobanu, Elena Santacroce, Domenico Lo Tartaro, Gianluca Degliesposti, Miriam D’Angerio, Maristella Leccese, Martina Cardi, Tommaso Trenti, Michela Cuccorese, and et al. 2024. "SARS-CoV-2 Vaccination Responses in Anti-CD20-Treated Progressive Multiple Sclerosis Patients Show Immunosenescence in Antigen-Specific B and T Cells" Vaccines 12, no. 8: 924. https://doi.org/10.3390/vaccines12080924