
Dose Considerations for Vaccinia Oncolytic Virus Based on Retrospective Reanalysis of Early and Late Clinical Trials

 ,
,
Abstract
:1. Introduction
2. Overview of PK and PD Following VOV Intratumoral Injection
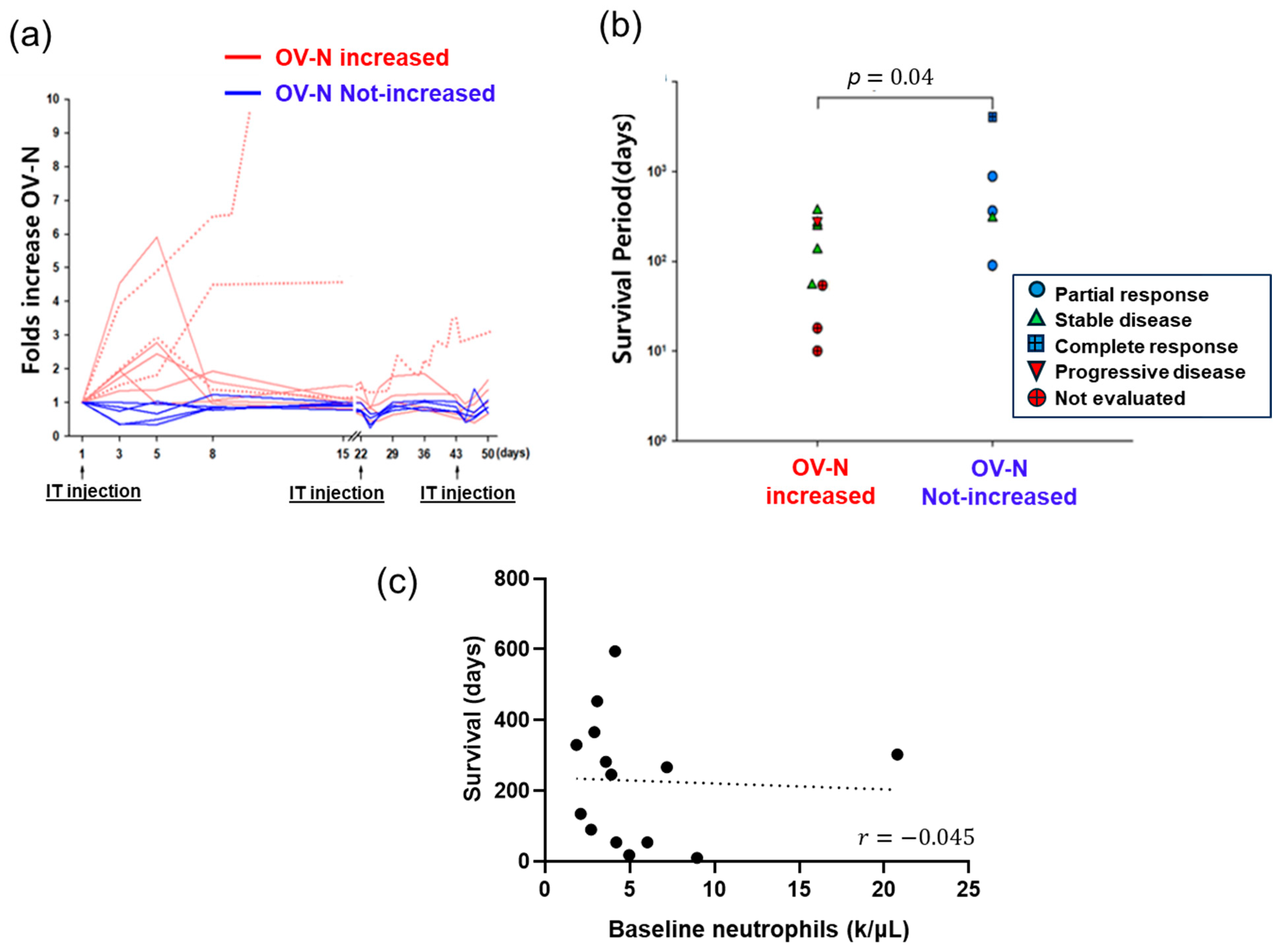
3. Retrospective Reanalysis of VOV Clinical Outcome
4. Proposed Hormetic Dose–Response
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, C.; Zhao, G.; Zhang, Z.; Yang, L.; Liu, S.; Li, G.; Wang, H.; Huang, J.; Wang, S.; Li, N. Immune Checkpoint Inhibitor-Associated Myocarditis: A Systematic Analysis of Case Reports. Front. Immunol. 2023, 14, 1275254. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef]
- Research, Center for Drug Evaluation and Research (CDER). Oncology (Cancer)/Hematologic Malignancies Approval Notifications. FDA, 2024. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/oncology-cancer-hematologic-malignancies-approval-notifications (accessed on 26 June 2024).
- News in Brief: First-in-Class T Cell Engager Approved for Lung Cancer. Nat. Biotechnol. 2024, 42, 824. [CrossRef] [PubMed]
- Mullard, A. FDA Approves First Tumour-Infiltrating Lymphocyte (TIL) Therapy, Bolstering Hopes for Cell Therapies in Solid Cancers. Nat. Rev. Drug Discov. 2024, 23, 238. [Google Scholar] [CrossRef] [PubMed]
- Soltantabar, P.; Lon, H.-K.; Parivar, K.; Wang, D.D.; Elmeliegy, M. Optimizing Benefit/Risk in Oncology: Review of Post-Marketing Dose Optimization and Reflections on the Road Ahead. Crit. Rev. Oncol. Hematol. 2023, 182, 103913. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined with Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic Virotherapy: Basic Principles, Recent Advances and Future Directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef]
- Melcher, A.; Harrington, K.; Vile, R. Oncolytic Virotherapy as Immunotherapy. Science 2021, 374, 1325–1326. [Google Scholar] [CrossRef]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic Virus Immunotherapy: Future Prospects for Oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic Virotherapy and Immunogenic Cancer Cell Death: Sharpening the Sword for Improved Cancer Treatment Strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef]
- Park, B.-H.; Hwang, T.; Liu, T.-C.; Sze, D.Y.; Kim, J.-S.; Kwon, H.-C.; Oh, S.Y.; Han, S.-Y.; Yoon, J.-H.; Hong, S.-H.; et al. Use of a Targeted Oncolytic Poxvirus, JX-594, in Patients with Refractory Primary or Metastatic Liver Cancer: A Phase I Trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.M.; Nieva, J.; Hwang, T.-H.; Moon, A.; Patt, R.; et al. Intravenous Delivery of a Multi-Mechanistic Cancer-Targeted Oncolytic Poxvirus in Humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized Dose-Finding Clinical Trial of Oncolytic Immunotherapeutic Vaccinia JX-594 in Liver Cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-Based Oncolytic Immunotherapy Pexastimogene Devacirepvec in Patients with Advanced Hepatocellular Carcinoma after Sorafenib Failure: A Randomized Multicenter Phase IIb Trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral Recombinant GM-CSF-Encoding Virus as Gene Therapy in Patients with Cutaneous Melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 Study of Intratumoral Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus, in Pediatric Cancer Patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.-H.; Sommermann, E.M.; Maruri Avidal, L.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef]
- Gaya, A.; Akle, C.A.; Mudan, S.; Grange, J. The Concept of Hormesis in Cancer Therapy—Is Less More? Cureus 2015, 7, e261. [Google Scholar] [CrossRef]
- Calabrese, E.J. Hormesis: Principles and Applications. Homeopathy 2015, 104, 69–82. [Google Scholar] [CrossRef]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic Armed Oncolytic and Immunologic Therapy for Cancer with JX-594, a Targeted Poxvirus Expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.-H.; et al. Oncolytic Vaccinia Virus Disrupts Tumor-Associated Vasculature in Humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Breitbach, C.J.; Moon, A.; Heo, J.; Lee, Y.K.; Cho, M.; Lee, J.W.; Kim, S.-G.; Kang, D.H.; Bell, J.C.; et al. Oncolytic and Immunotherapeutic Vaccinia Induces Antibody-Mediated Complement-Dependent Cancer Cell Lysis in Humans. Sci. Transl. Med. 2013, 5, 185ra63. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.-H.; Moon, A.; Burke, J.; Ribas, A.; Stephenson, J.; Breitbach, C.J.; Daneshmand, M.; De Silva, N.; Parato, K.; Diallo, J.-S.; et al. A Mechanistic Proof-of-Concept Clinical Trial With JX-594, a Targeted Multi-Mechanistic Oncolytic Poxvirus, in Patients with Metastatic Melanoma. Mol. Ther. 2011, 19, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Erinjeri, J.; Heo, J.; Borad, M.J.; Luca, A.; Burke, J.; Pelusio, A.; Agathon, D.; et al. PHOCUS: A Phase 3, Randomized, Open-Label Study of Sequential Treatment with Pexa-Vec (JX-594) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2023, 13, 256–272. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Baldwin, L.A. Toxicology Rethinks Its Central Belief. Nature 2003, 421, 691–692. [Google Scholar] [CrossRef]
- Pearce, O.M.; Läubli, H.; Bui, J.; Varki, A. Hormesis in Cancer Immunology: Does the Quantity of an Immune Reactant Matter? Oncoimmunology 2014, 3, e29312. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H.; Verhagen, A.; Secrest, P.; Zhang, J.; Varki, N.M.; Crocker, P.R.; Bui, J.D.; Varki, A. Inverse Hormesis of Cancer Growth Mediated by Narrow Ranges of Tumor-Directed Antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 5998–6003. [Google Scholar] [CrossRef]
- Cui, J.; Yang, G.; Pan, Z.; Zhao, Y.; Liang, X.; Li, W.; Cai, L. Hormetic Response to Low-Dose Radiation: Focus on the Immune System and Its Clinical Implications. Int. J. Mol. Sci. 2017, 18, 280. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Giordano, J.J.; Kozumbo, W.J.; Leak, R.K.; Bhatia, T.N. Hormesis Mediates Dose-Sensitive Shifts in Macrophage Activation Patterns. Pharmacol. Res. 2018, 137, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Li, S.; Tang, B.; Wang, X.; Xiao, Y.; Cheke, R.A. Hormetic and Synergistic Effects of Cancer Treatments Revealed by Modelling Combinations of Radio—Or Chemotherapy with Immunotherapy. BMC Cancer 2023, 23, 1040. [Google Scholar] [CrossRef]
- Zahid, K.R.; Raza, U.; Tumbath, S.; Jiang, L.; Xu, W.; Huang, X. Neutrophils: Musketeers against Immunotherapy. Front. Oncol. 2022, 12, 975981. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Bell, J.C.; Hwang, T.-H.; Kirn, D.H.; Burke, J. The Emerging Therapeutic Potential of the Oncolytic Immunotherapeutic Pexa-Vec (JX-594). Oncolytic Virother. 2015, 4, 25–31. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef]
- Emmons, T.R.; Giridharan, T.; Singel, K.L.; Khan, A.N.H.; Ricciuti, J.; Howard, K.; Silva-Del Toro, S.L.; Debreceni, I.L.; Aarts, C.E.M.; Brouwer, M.C.; et al. Mechanisms Driving Neutrophil-Induced T-Cell Immunoparalysis in Ovarian Cancer. Cancer Immunol. Res. 2021, 9, 790–810. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting Myeloid-Derived Suppressor Cell Trafficking Enhances T Cell Immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef]
- Le Louedec, F.; Leenhardt, F.; Marin, C.; Chatelut, É.; Evrard, A.; Ciccolini, J. Cancer Immunotherapy Dosing: A Pharmacokinetic/Pharmacodynamic Perspective. Vaccines 2020, 8, 632. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, M.A.; Luker, K.E.; Sonstein, J.; Núñez, G.; Curtis, J.L.; Luker, G.D. Protective Effect of Toll-like Receptor 4 in Pulmonary Vaccinia Infection. PLoS Pathog. 2008, 4, e1000153. [Google Scholar] [CrossRef]
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I.; et al. Phase I/II Study of PexaVec in Combination with Immune Checkpoint Inhibition in Refractory Metastatic Colorectal Cancer. J. Immunother. Cancer 2023, 11, e005640. [Google Scholar] [CrossRef] [PubMed]
- Rha, S.Y.; Bae, W.K.; Kim, C.; Oh, S.Y.; Lee, H.W.; Park, K.; Mar, N.; Kichenadasse, G.; Ha, H.K.; Pachynski, R.; et al. 1885P Pexa-Vec (Thymidine Kinase-Deactivated Vaccinia Virus plus GM-CSF) in Combination with Cemiplimab (REGN2810; ANTI-PD-1) for Metastatic or Unresectable Renal Cell Carcinoma REN026: Results from a Phase II Study. Ann. Oncol. 2023, 34, S1013–S1014. [Google Scholar] [CrossRef]
- Islam, S.M.B.U.; Hong, Y.M.; Ornella, M.S.C.; Ngabire, D.; Jang, H.; Cho, E.; Kim, E.-K.; Hale, J.J.J.; Kim, C.H.; Ahn, S.C.; et al. Engineering and Preclinical Evaluation of Western Reserve Oncolytic Vaccinia Virus Expressing A167Y Mutant Herpes Simplex Virus Thymidine Kinase. Biomedicines 2020, 8, 426. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Disease | No. of Pts * | Dose/Cycle | Primary Objective | Conclusion | Ref. |
|---|---|---|---|---|---|---|
| Proof of concept | Melanoma | 7 | 1 × 104~8 × 107 pfu (i.t.)/twice weekly, for 6 weeks | Evaluation of safety and efficacy of Pexa-Vec | Injected and non-injected tumor lesions regressed Toxicity was minimal Tumor response: 71.4% | [20] |
| 1 Dose-escalation (NCT00629759) | Primary/metastatic liver cancer | 14 | 1 × 108, 3 × 108, 1 × 109, 3 × 109 pfu (i.t.)/4 cycles (every 3 weeks) | Evaluation of safety of Pexa-Vec Determine MTD and/or MFD | MTD of 109 pfu was confirmed; mild-to moderate flu-like symptoms appeared as the most common adverse event Tumor response: 21.4% | [16] |
| 2a Randomized dose-finding (NCT00554372) | HCC | 28 | 1 × 108, 1 × 109 pfu (i.t.)/3 cycles (every 2 weeks) | Proportion of subjects achieving disease control (non-progressive disease) at 8 weeks after initiation of treatment | Survival duration was significantly dose-related, in contrast to tumor response and immune endpoints Tumor response: 14.3% | [18] |
| 2b (TRAVERSE) (NCT01387555) | HCC | 65 | 1 × 109 pfu (i.v.)/day 1 f/b 1 × 109 pfu (i.t.)/5 cycles (day 8, week 3, 6, 12, and 18) | Overall survival (OS) | Pexa-Vec did not improve OS as second-line therapy after sorafenib failure Tumor response: 0.0% | [19] |
| Dose a | Tumor Response (RECIST) b %(n) | Disease | Original Description on Systemic Toxicity | Investigators’ Retrospective Reanalysis | Ref. |
|---|---|---|---|---|---|
| 1 × 104–8 × 107 | 71.4% (5/7 pts) | Melanoma | Flu-like symptom with ≥4 × 107 pfu, 12% No hematological and ANC level change | N/A | [20] |
| 1 × 108–3 × 108 | 26.3% (5/19 pts) | Lung/thymic, colon, gastric, liver, extragonadal | Mild flu-like symptoms (grade 1–2) No grade 4–5 Adverse effect | Tumor response is associated with ANC decrease post treatment Adaptive tumor immunity was confirmed in long-time survivors with complete response | [16,18,27] |
| 1 × 109–3 × 109 | 1.1% (1/88 pts) | Renal, colon, liver, melanoma | Treatment-related DLT
| DLT may be associated with severe systemic inflammation following sustained ANC increase post treatment | [16,18,19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cyrelle Ornella, M.S.; Kim, J.-J.; Cho, E.; Cho, M.; Hwang, T.-H. Dose Considerations for Vaccinia Oncolytic Virus Based on Retrospective Reanalysis of Early and Late Clinical Trials. Vaccines 2024, 12, 1010. https://doi.org/10.3390/vaccines12091010
Cyrelle Ornella MS, Kim J-J, Cho E, Cho M, Hwang T-H. Dose Considerations for Vaccinia Oncolytic Virus Based on Retrospective Reanalysis of Early and Late Clinical Trials. Vaccines. 2024; 12(9):1010. https://doi.org/10.3390/vaccines12091010
Chicago/Turabian StyleCyrelle Ornella, Mefotse Saha, Jae-Joon Kim, Euna Cho, Mong Cho, and Tae-Ho Hwang. 2024. "Dose Considerations for Vaccinia Oncolytic Virus Based on Retrospective Reanalysis of Early and Late Clinical Trials" Vaccines 12, no. 9: 1010. https://doi.org/10.3390/vaccines12091010