
Systemic and Mucosal Antibody Responses to SARS-CoV-2 Variant-Specific Prime-and-Boost and Prime-and-Spike Vaccination: A Comparison of Intramuscular and Intranasal Bivalent Vaccine Administration in a Murine Model
 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. Mice Immunization and Sample Collection
2.2. SARS-CoV-2-Specific Antibody Measurements
2.3. Surrogate Neutralization ELISA (snELISA) Assay to Evaluate Neutralization Activity in Serum and BALF Samples
2.4. Principal Component Analysis of Neutralization and Serology Data
3. Results
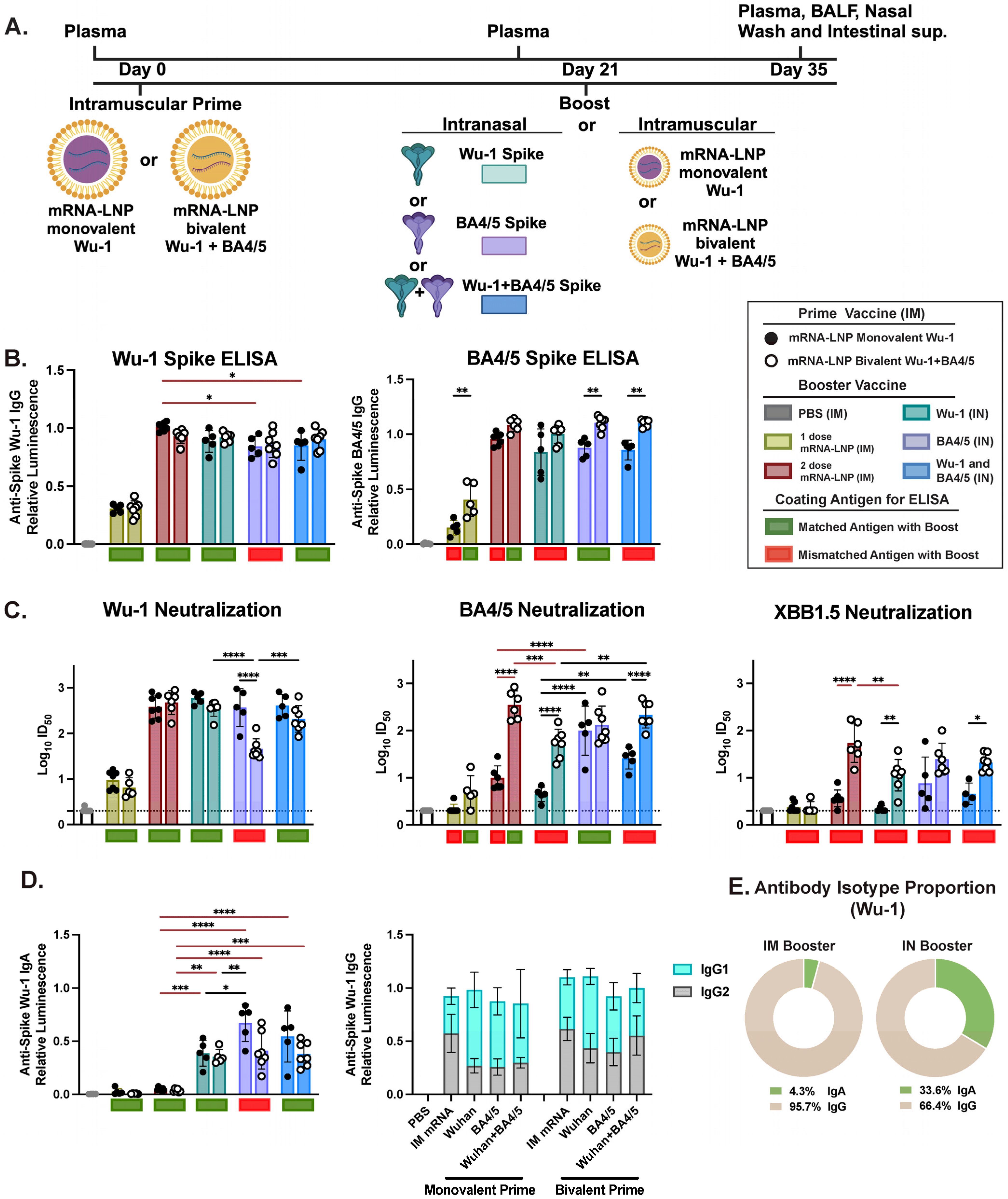
3.1. IM Prime Shapes Systemic Humoral Responses, Primarily Driven by Variant-Specific Neutralizing Responses
3.2. Bivalent mRNA-LNP IM Prime Broadens Neutralizing Responses in Serum
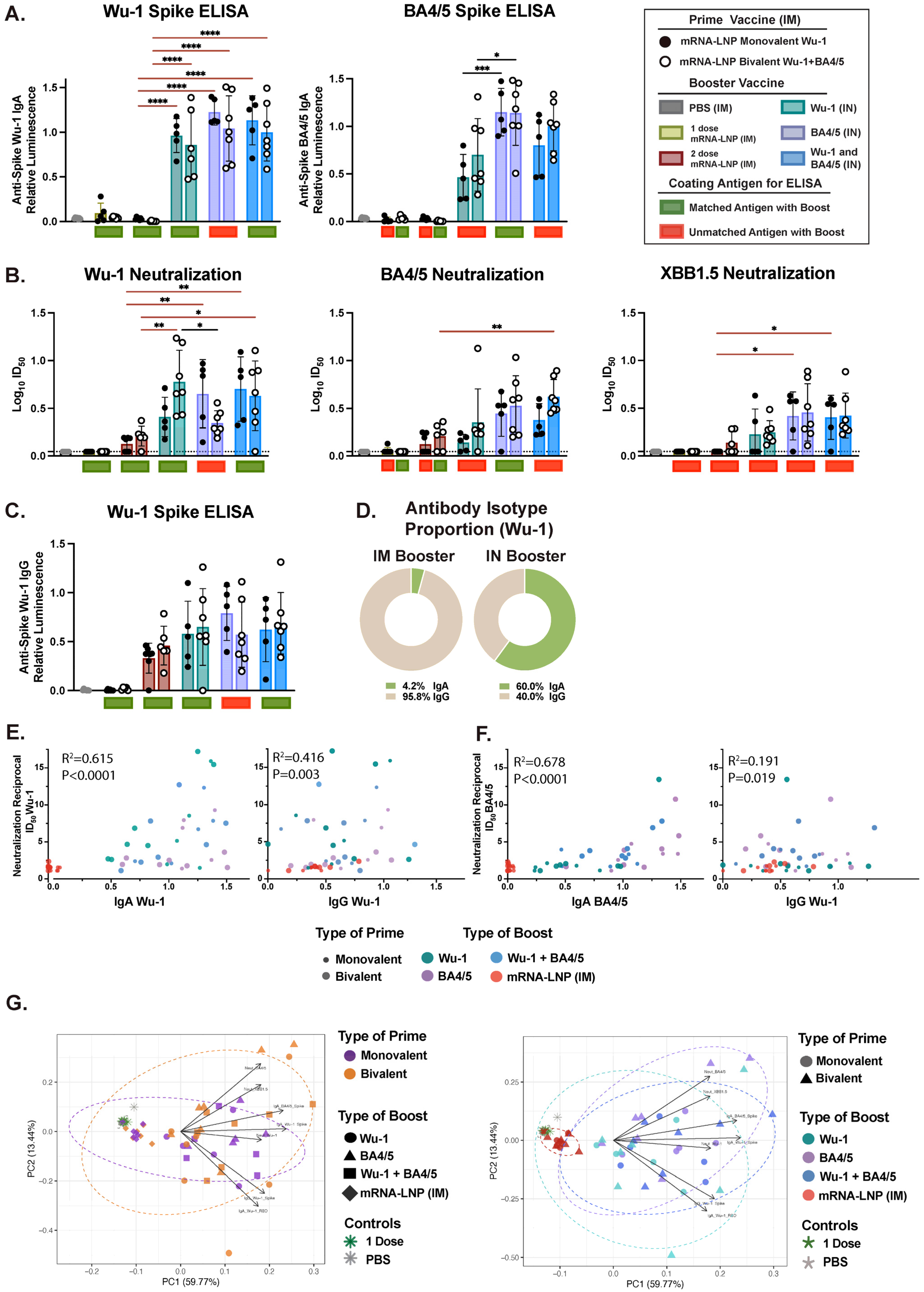
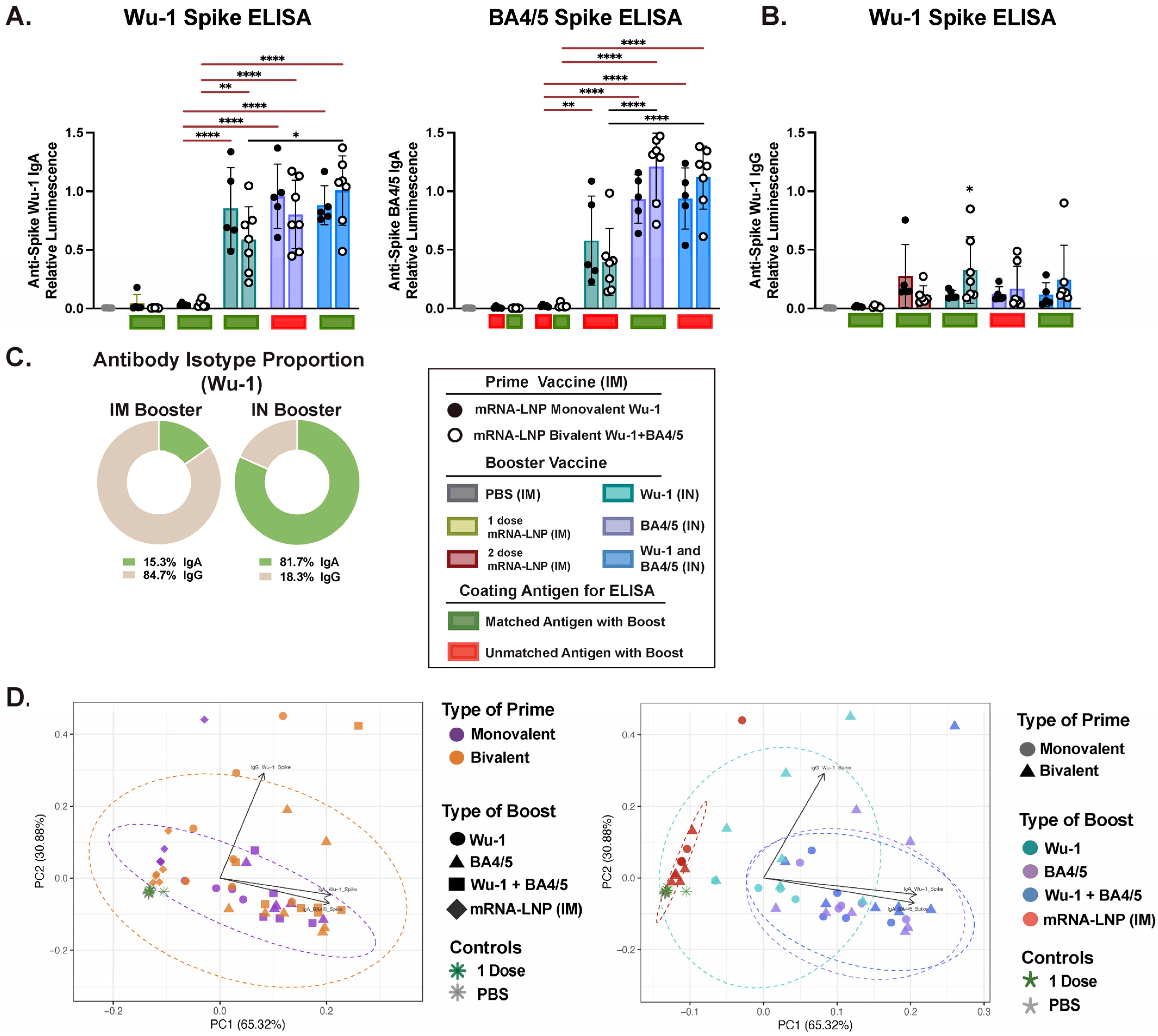
3.3. Variant-Specific IN Vaccines Induce Distinct Antigen-Specific IgA Responses in Nasal and BAL Biofluids
3.4. IM and IN Vaccination Induce Potent IgG Responses in the Intestinal Lumen
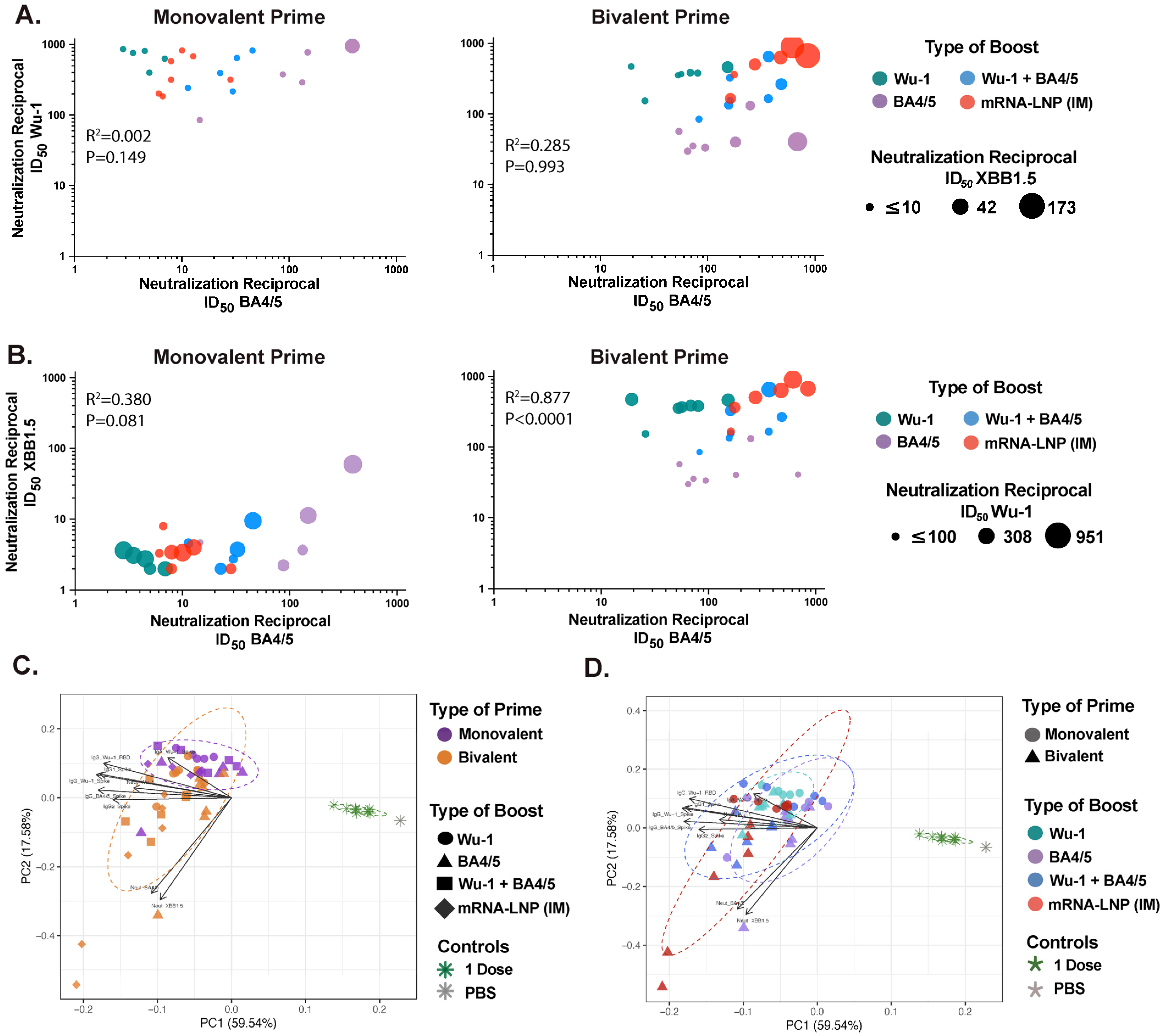
3.5. Effect of Administration Route on Humoral Response Profiles and Antibody Correlations Across Compartments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.; et al. SARS-CoV-2 Omicron has extensive but incomplete escape of Pfizer BNT162b2 elicited neutralization and requires ACE2 for infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Chemaitelly, H.; Tang, P.; Hasan, M.R.; AlMukdad, S.; Yassine, H.M.; Benslimane, F.M.; Al Khatib, H.A.; Coyle, P.; Ayoub, H.H.; Al Kanaani, Z.; et al. Waning of BNT162b2 Vaccine Protection against SARS-CoV-2 Infection in Qatar. N. Engl. J. Med. 2021, 385, e83. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e49. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Addetia, A.; Seo, A.J.; Brown, J.; Sprouse, K.; Logue, J.; Clark, E.; Franko, N.; Chu, H.; Veesler, D. Persistent immune imprinting occurs after vaccination with the COVID-19 XBB.1.5 mRNA booster in humans. Immunity 2024, 57, 904–911.e4. [Google Scholar] [CrossRef]
- Tan, S.T.; Kwan, A.T.; Rodriguez-Barraquer, I.; Singer, B.J.; Park, H.J.; Lewnard, J.A.; Sears, D.; Lo, N.C. Infectiousness of SARS-CoV-2 breakthrough infections and reinfections during the Omicron wave. Nat. Med. 2023, 29, 358–365. [Google Scholar] [CrossRef]
- Regenhardt, E.; Kirsten, H.; Weiss, M.; Lubbert, C.; Stehr, S.N.; Remane, Y.; Pietsch, C.; Honemann, M.; von Braun, A. SARS-CoV-2 Vaccine Breakthrough Infections of Omicron and Delta Variants in Healthcare Workers. Vaccines 2023, 11, 958. [Google Scholar] [CrossRef]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B.1.617.2) variant in vaccinated and unvaccinated individuals in the UK: A prospective, longitudinal, cohort study. Lancet Infect. Dis. 2022, 22, 183–195. [Google Scholar] [CrossRef]
- Chu, L.; Vrbicky, K.; Montefiori, D.; Huang, W.; Nestorova, B.; Chang, Y.; Carfi, A.; Edwards, D.K.; Oestreicher, J.; Legault, H.; et al. Immune response to SARS-CoV-2 after a booster of mRNA-1273: An open-label phase 2 trial. Nat. Med. 2022, 28, 1042–1049. [Google Scholar] [CrossRef]
- Kaku, C.I.; Bergeron, A.J.; Ahlm, C.; Normark, J.; Sakharkar, M.; Forsell, M.N.E.; Walker, L.M. Recall of preexisting cross-reactive B cell memory after Omicron BA.1 breakthrough infection. Sci. Immunol. 2022, 7, eabq3511. [Google Scholar] [CrossRef]
- Kuhlmann, C.; Mayer, C.K.; Claassen, M.; Maponga, T.; Burgers, W.A.; Keeton, R.; Riou, C.; Sutherland, A.D.; Suliman, T.; Shaw, M.L.; et al. Breakthrough infections with SARS-CoV-2 omicron despite mRNA vaccine booster dose. Lancet 2022, 399, 625–626. [Google Scholar] [CrossRef]
- Lange, B.; Gerigk, M.; Tenenbaum, T. Breakthrough Infections in BNT162b2-Vaccinated Health Care Workers. N. Engl. J. Med. 2021, 385, 1145–1146. [Google Scholar] [CrossRef] [PubMed]
- Kustin, T.; Harel, N.; Finkel, U.; Perchik, S.; Harari, S.; Tahor, M.; Caspi, I.; Levy, R.; Leshchinsky, M.; Ken Dror, S.; et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2-mRNA-vaccinated individuals. Nat. Med. 2021, 27, 1379–1384. [Google Scholar] [CrossRef]
- Bergwerk, M.; Gonen, T.; Lustig, Y.; Amit, S.; Lipsitch, M.; Cohen, C.; Mandelboim, M.; Levin, E.G.; Rubin, C.; Indenbaum, V.; et al. COVID-19 Breakthrough Infections in Vaccinated Health Care Workers. N. Engl. J. Med. 2021, 385, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.J.; Perez Marc, G.; Gobeil, P.; Diaz, R.S.; Heizer, G.; Llapur, C.; Makarkov, A.I.; Vasconcellos, E.; Pillet, S.; Riera, F.; et al. Efficacy and Safety of a Recombinant Plant-Based Adjuvanted COVID-19 Vaccine. N. Engl. J. Med. 2022, 386, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Moreira, E.D., Jr.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Self, W.H.; Adams, K.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Association Between mRNA Vaccination and COVID-19 Hospitalization and Disease Severity. JAMA 2021, 326, 2043–2054. [Google Scholar] [CrossRef]
- Menni, C.; May, A.; Polidori, L.; Louca, P.; Wolf, J.; Capdevila, J.; Hu, C.; Ourselin, S.; Steves, C.J.; Valdes, A.M.; et al. COVID-19 vaccine waning and effectiveness and side-effects of boosters: A prospective community study from the ZOE COVID Study. Lancet Infect. Dis. 2022, 22, 1002–1010. [Google Scholar] [CrossRef]
- Lucas, C.; Vogels, C.B.F.; Yildirim, I.; Rothman, J.E.; Lu, P.; Monteiro, V.; Gehlhausen, J.R.; Campbell, M.; Silva, J.; Tabachnikova, A.; et al. Impact of circulating SARS-CoV-2 variants on mRNA vaccine-induced immunity. Nature 2021, 600, 523–529. [Google Scholar] [CrossRef]
- Weber, T.; Dahling, S.; Rose, S.; Affeldt, P.; Vanshylla, K.; Ullrich, L.; Gieselmann, L.; Teipel, F.; Gruell, H.; Di Cristanziano, V.; et al. Enhanced SARS-CoV-2 humoral immunity following breakthrough infection builds upon the preexisting memory B cell pool. Sci. Immunol. 2023, 8, eadk5845. [Google Scholar] [CrossRef]
- Kemp, S.A.; Cheng, M.T.K.; Hamilton, W.L.; Kamelian, K.; Indian, S.-C.-G.C.; Singh, S.; Rakshit, P.; Agrawal, A.; Illingworth, C.J.R.; Gupta, R.K. Transmission of B.1.617.2 Delta variant between vaccinated healthcare workers. Sci. Rep. 2022, 12, 10492. [Google Scholar] [CrossRef]
- Tang, J.; Zeng, C.; Cox, T.M.; Li, C.; Son, Y.M.; Cheon, I.S.; Wu, Y.; Behl, S.; Taylor, J.J.; Chakraborty, R.; et al. Respiratory mucosal immunity against SARS-CoV-2 following mRNA vaccination. Sci. Immunol. 2022, 7, eadd4853. [Google Scholar] [CrossRef] [PubMed]
- Sheikh-Mohamed, S.; Isho, B.; Chao, G.Y.C.; Zuo, M.; Cohen, C.; Lustig, Y.; Nahass, G.R.; Salomon-Shulman, R.E.; Blacker, G.; Fazel-Zarandi, M.; et al. Systemic and mucosal IgA responses are variably induced in response to SARS-CoV-2 mRNA vaccination and are associated with protection against subsequent infection. Mucosal Immunol. 2022, 15, 799–808. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar] [CrossRef]
- Bartsch, Y.C.; Wang, C.; Zohar, T.; Fischinger, S.; Atyeo, C.; Burke, J.S.; Kang, J.; Edlow, A.G.; Fasano, A.; Baden, L.R.; et al. Humoral signatures of protective and pathological SARS-CoV-2 infection in children. Nat. Med. 2021, 27, 454–462. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Demone, J.; Maltseva, M.; Nourimand, M.; Nasr-Sharif, M.; Galipeau, Y.; Alarcon, E.I.; Langlois, M.A.; MacLean, A.M. Scalable agroinfiltration-based production of SARS-CoV-2 antigens for use in diagnostic assays and subunit vaccines. PLoS ONE 2022, 17, e0277668. [Google Scholar] [CrossRef]
- Chalkias, S.; Whatley, J.L.; Eder, F.; Essink, B.; Khetan, S.; Bradley, P.; Brosz, A.; McGhee, N.; Tomassini, J.E.; Chen, X.; et al. Original SARS-CoV-2 monovalent and Omicron BA.4/BA.5 bivalent COVID-19 mRNA vaccines: Phase 2/3 trial interim results. Nat. Med. 2023, 29, 2325–2333. [Google Scholar] [CrossRef]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023, 29, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, S.; Harper, C.; Vrbicky, K.; Walsh, S.R.; Essink, B.; Brosz, A.; McGhee, N.; Tomassini, J.E.; Chen, X.; Chang, Y.; et al. A Bivalent Omicron-Containing Booster Vaccine against COVID-19. N. Engl. J. Med. 2022, 387, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Davis-Gardner, M.E.; Lai, L.; Wali, B.; Samaha, H.; Solis, D.; Lee, M.; Porter-Morrison, A.; Hentenaar, I.T.; Yamamoto, F.; Godbole, S.; et al. Neutralization against BA.2.75.2, BQ.1.1, and XBB from mRNA Bivalent Booster. N. Engl. J. Med. 2023, 388, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Kurhade, C.; Patel, S.; Kitchin, N.; Tompkins, K.; Cutler, M.; Cooper, D.; Yang, Q.; Cai, H.; Muik, A.; et al. Neutralization of BA.4-BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with Bivalent Vaccine. N. Engl. J. Med. 2023, 388, 854–857. [Google Scholar] [CrossRef]
- Collier, A.Y.; Miller, J.; Hachmann, N.P.; McMahan, K.; Liu, J.; Bondzie, E.A.; Gallup, L.; Rowe, M.; Schonberg, E.; Thai, S.; et al. Immunogenicity of BA.5 Bivalent mRNA Vaccine Boosters. N. Engl. J. Med. 2023, 388, 565–567. [Google Scholar] [CrossRef]
- Wang, Q.; Bowen, A.; Valdez, R.; Gherasim, C.; Gordon, A.; Liu, L.; Ho, D.D. Antibody Response to Omicron BA.4-BA.5 Bivalent Booster. N. Engl. J. Med. 2023, 388, 567–569. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Tam, A.R.; Valdez, R.; Gordon, A.; Liu, L.; Ho, D.D. Deep immunological imprinting due to the ancestral spike in the current bivalent COVID-19 vaccine. Cell Rep. Med. 2023, 4, 101258. [Google Scholar] [CrossRef]
- Wang, Q.; Bowen, A.; Tam, A.R.; Valdez, R.; Stoneman, E.; Mellis, I.A.; Gordon, A.; Liu, L.; Ho, D.D. SARS-CoV-2 neutralising antibodies after bivalent versus monovalent booster. Lancet Infect. Dis. 2023, 23, 527–528. [Google Scholar] [CrossRef]
- Wang, Q.; Bowen, A.; Ho, J.; Zhang, R.M.; Valdez, R.; Stoneman, E.; Gordon, A.; Liu, L.; Ho, D.D. SARS-CoV-2 neutralising antibodies after a second BA.5 bivalent booster. Lancet 2023, 402, 1827–1828. [Google Scholar] [CrossRef]
- Johnston, T.S.; Li, S.H.; Painter, M.M.; Atkinson, R.K.; Douek, N.R.; Reeg, D.B.; Douek, D.C.; Wherry, E.J.; Hensley, S.E. Immunological imprinting shapes the specificity of human antibody responses against SARS-CoV-2 variants. Immunity 2024, 57, 912–925.e914. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Bowen, A.; Mellis, I.A.; Valdez, R.; Gherasim, C.; Gordon, A.; Liu, L.; Ho, D.D. XBB.1.5 monovalent mRNA vaccine booster elicits robust neutralizing antibodies against XBB subvariants and JN.1. Cell Host Microbe 2024, 32, 315–321.e313. [Google Scholar] [CrossRef]
- Altarawneh, H.N.; Chemaitelly, H.; Ayoub, H.H.; Tang, P.; Hasan, M.R.; Yassine, H.M.; Al-Khatib, H.A.; Al Thani, A.A.; Coyle, P.; Al-Kanaani, Z.; et al. Effects of previous infection, vaccination, and hybrid immunity against symptomatic Alpha, Beta, and Delta SARS-CoV-2 infections: An observational study. EBioMedicine 2023, 95, 104734. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bretones, M.; Fouchier, R.A.; Koopmans, M.P.; van Nierop, G.P. Impact of antigenic evolution and original antigenic sin on SARS-CoV-2 immunity. J. Clin. Investig. 2023, 133, e162192. [Google Scholar] [CrossRef]
- Lin, D.Y.; Xu, Y.; Gu, Y.; Zeng, D.; Sunny, S.K.; Moore, Z. Durability of Bivalent Boosters against Omicron Subvariants. N. Engl. J. Med. 2023, 388, 1818–1820. [Google Scholar] [CrossRef]
- Han, A.X.; de Jong, S.P.J.; Russell, C.A. Co-evolution of immunity and seasonal influenza viruses. Nat. Rev. Microbiol. 2023, 21, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, M.; Keeshan, A.; Cooper, C.; Langlois, M.A. Immune imprinting: The persisting influence of the first antigenic encounter with rapidly evolving viruses. Hum. Vaccin. Immunother. 2024, 20, 2384192. [Google Scholar] [CrossRef]
- Koutsakos, M.; Ellebedy, A.H. Immunological imprinting: Understanding COVID-19. Immunity 2023, 56, 909–913. [Google Scholar] [CrossRef]
- Huang, C.Q.; Vishwanath, S.; Carnell, G.W.; Chan, A.C.Y.; Heeney, J.L. Immune imprinting and next-generation coronavirus vaccines. Nat. Microbiol. 2023, 8, 1971–1985. [Google Scholar] [CrossRef]
- Carreno, J.M.; Singh, G.; Simon, V.; Krammer, F.; PVI Study Group. Bivalent COVID-19 booster vaccines and the absence of BA.5-specific antibodies. Lancet Microbe 2023, 4, e569. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Pinto, D.; Walls, A.C.; Liu, Z.; De Marco, A.; Benigni, F.; Zatta, F.; Silacci-Fregni, C.; Bassi, J.; Sprouse, K.R.; et al. Imprinted antibody responses against SARS-CoV-2 Omicron sublineages. Science 2022, 378, 619–627. [Google Scholar] [CrossRef]
- Roltgen, K.; Nielsen, S.C.A.; Silva, O.; Younes, S.F.; Zaslavsky, M.; Costales, C.; Yang, F.; Wirz, O.F.; Solis, D.; Hoh, R.A.; et al. Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell 2022, 185, 1025–1040.e1014. [Google Scholar] [CrossRef] [PubMed]
- Aydillo, T.; Rombauts, A.; Stadlbauer, D.; Aslam, S.; Abelenda-Alonso, G.; Escalera, A.; Amanat, F.; Jiang, K.; Krammer, F.; Carratala, J.; et al. Immunological imprinting of the antibody response in COVID-19 patients. Nat. Commun. 2021, 12, 3781. [Google Scholar] [CrossRef]
- Marking, U.; Bladh, O.; Havervall, S.; Svensson, J.; Greilert-Norin, N.; Aguilera, K.; Kihlgren, M.; Salomonsson, A.C.; Mansson, M.; Gallini, R.; et al. 7-month duration of SARS-CoV-2 mucosal immunoglobulin-A responses and protection. Lancet Infect. Dis. 2023, 23, 150–152. [Google Scholar] [CrossRef]
- Sheikh-Mohamed, S.; Sanders, E.C.; Gommerman, J.L.; Tal, M.C. Guardians of the oral and nasopharyngeal galaxy: IgA and protection against SARS-CoV-2 infection. Immunol. Rev. 2022, 309, 75–85. [Google Scholar] [CrossRef]
- Havervall, S.; Marking, U.; Svensson, J.; Greilert-Norin, N.; Bacchus, P.; Nilsson, P.; Hober, S.; Gordon, M.; Blom, K.; Klingstrom, J.; et al. Anti-Spike Mucosal IgA Protection against SARS-CoV-2 Omicron Infection. N. Engl. J. Med. 2022, 387, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Bhavsar, D.; Singh, G.; Floda, D.; Srivastava, K.; Gleason, C.; Group, P.S.; Carreno, J.M.; Simon, V.; Krammer, F. SARS-CoV-2 vaccination induces mucosal antibody responses in previously infected individuals. Nat. Commun. 2022, 13, 5135. [Google Scholar] [CrossRef]
- Bhavsar, D.; Singh, G.; Sano, K.; Gleason, C.; Srivastava, K.; Group, P.S.; Carreno, J.M.; Simon, V.; Krammer, F. Mucosal antibody responses to SARS-CoV-2 booster vaccination and breakthrough infection. mBio 2023, 14, e0228023. [Google Scholar] [CrossRef]
- Stark, F.C.; Akache, B.; Deschatelets, L.; Tran, A.; Stuible, M.; Durocher, Y.; McCluskie, M.J.; Agbayani, G.; Dudani, R.; Harrison, B.A.; et al. Intranasal immunization with a proteosome-adjuvanted SARS-CoV-2 spike protein-based vaccine is immunogenic and efficacious in mice and hamsters. Sci. Rep. 2022, 12, 9772. [Google Scholar] [CrossRef]
- Hassan, A.O.; Feldmann, F.; Zhao, H.; Curiel, D.T.; Okumura, A.; Tang-Huau, T.L.; Case, J.B.; Meade-White, K.; Callison, J.; Lovaglio, J.; et al. A single intranasal dose of chimpanzee adenovirus-vectored vaccine protects against SARS-CoV-2 infection in rhesus macaques. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Bricker, T.L.; Darling, T.L.; Hassan, A.O.; Harastani, H.H.; Soung, A.; Jiang, X.; Dai, Y.N.; Zhao, H.; Adams, L.J.; Holtzman, M.J.; et al. A single intranasal or intramuscular immunization with chimpanzee adenovirus-vectored SARS-CoV-2 vaccine protects against pneumonia in hamsters. Cell Rep. 2021, 36, 109400. [Google Scholar] [CrossRef]
- Langel, S.N.; Johnson, S.; Martinez, C.I.; Tedjakusuma, S.N.; Peinovich, N.; Dora, E.G.; Kuehl, P.J.; Irshad, H.; Barrett, E.G.; Werts, A.; et al. Adenovirus type 5 SARS-CoV-2 vaccines delivered orally or intranasally reduced disease severity and transmission in a hamster model. Sci. Transl. Med. 2022, 14, eabn6868. [Google Scholar] [CrossRef]
- Afkhami, S.; D’Agostino, M.R.; Zhang, A.; Stacey, H.D.; Marzok, A.; Kang, A.; Singh, R.; Bavananthasivam, J.; Ye, G.; Luo, X.; et al. Respiratory mucosal delivery of next-generation COVID-19 vaccine provides robust protection against both ancestral and variant strains of SARS-CoV-2. Cell 2022, 185, 896–915.e819. [Google Scholar] [CrossRef]
- Du, Y.; Xu, Y.; Feng, J.; Hu, L.; Zhang, Y.; Zhang, B.; Guo, W.; Mai, R.; Chen, L.; Fang, J.; et al. Intranasal administration of a recombinant RBD vaccine induced protective immunity against SARS-CoV-2 in mouse. Vaccine 2021, 39, 2280–2287. [Google Scholar] [CrossRef]
- Maltseva, M.; Galipeau, Y.; Renner, T.M.; Deschatelets, L.; Durocher, Y.; Akache, B.; Langlois, M.A. Characterization of Systemic and Mucosal Humoral Immune Responses to an Adjuvanted Intranasal SARS-CoV-2 Protein Subunit Vaccine Candidate in Mice. Vaccines 2022, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.F.; Calderon, K.; Rojas-Neyra, A.; Vakharia, V.N.; Choque-Guevara, R.; Montalvan-Avalos, A.; Poma-Acevedo, A.; Rios-Matos, D.; Agurto-Arteaga, A.; Cauti-Mendoza, M.G.; et al. Intranasal vaccination of hamsters with a Newcastle disease virus vector expressing the S1 subunit protects animals against SARS-CoV-2 disease. Sci. Rep. 2022, 12, 10359. [Google Scholar] [CrossRef]
- van Doremalen, N.; Purushotham, J.N.; Schulz, J.E.; Holbrook, M.G.; Bushmaker, T.; Carmody, A.; Port, J.R.; Yinda, C.K.; Okumura, A.; Saturday, G.; et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci. Transl. Med. 2021, 13, eabh0755. [Google Scholar] [CrossRef] [PubMed]
- King, R.G.; Silva-Sanchez, A.; Peel, J.N.; Botta, D.; Dickson, A.M.; Pinto, A.K.; Meza-Perez, S.; Allie, S.R.; Schultz, M.D.; Liu, M.; et al. Single-Dose Intranasal Administration of AdCOVID Elicits Systemic and Mucosal Immunity against SARS-CoV-2 and Fully Protects Mice from Lethal Challenge. Vaccines 2021, 9, 881. [Google Scholar] [CrossRef]
- Lapuente, D.; Fuchs, J.; Willar, J.; Vieira Antao, A.; Eberlein, V.; Uhlig, N.; Issmail, L.; Schmidt, A.; Oltmanns, F.; Peter, A.S.; et al. Protective mucosal immunity against SARS-CoV-2 after heterologous systemic prime-mucosal boost immunization. Nat. Commun. 2021, 12, 6871. [Google Scholar] [CrossRef]
- Hassan, A.O.; Kafai, N.M.; Dmitriev, I.P.; Fox, J.M.; Smith, B.K.; Harvey, I.B.; Chen, R.E.; Winkler, E.S.; Wessel, A.W.; Case, J.B.; et al. A Single-Dose Intranasal ChAd Vaccine Protects Upper and Lower Respiratory Tracts against SARS-CoV-2. Cell 2020, 183, 169–184.e113. [Google Scholar] [CrossRef] [PubMed]
- Agbayani, G.; Akache, B.; Renner, T.M.; Tran, A.; Stuible, M.; Dudani, R.; Harrison, B.A.; Duque, D.; Bavananthasivam, J.; Deschatelets, L.; et al. Intranasal administration of unadjuvanted SARS-CoV-2 spike antigen boosts antigen-specific immune responses induced by parenteral protein subunit vaccine prime in mice and hamsters. Eur. J. Immunol. 2024, 54, e2350620. [Google Scholar] [CrossRef]
- Mao, T.; Israelow, B.; Suberi, A.; Zhou, L.; Reschke, M.; Pena-Hernandez, M.A.; Dong, H.; Homer, R.J.; Saltzman, W.M.; Iwasaki, A. Unadjuvanted intranasal spike vaccine booster elicits robust protective mucosal immunity against sarbecoviruses. Science 2022, 378, eabo2523. [Google Scholar] [CrossRef]
- Christensen, D.; Polacek, C.; Sheward, D.J.; Hanke, L.; Moliner-Morro, A.; McInerney, G.; Murrell, B.; Hartmann, K.T.; Jensen, H.E.; Jungersen, G.; et al. Protection against SARS-CoV-2 transmission by a parenteral prime-Intranasal boost vaccine strategy. EBioMedicine 2022, 84, 104248. [Google Scholar] [CrossRef] [PubMed]
- Beitari, S.; Agbayani, G.; Hewitt, M.; Duque, D.; Bavananthasivam, J.; Sandhu, J.K.; Akache, B.; Hadzisejdic, I.; Tran, A. Effectiveness of VSV vectored SARS-CoV-2 spike when administered through intranasal, intramuscular or a combination of both. Sci. Rep. 2023, 13, 21390. [Google Scholar] [CrossRef] [PubMed]
- Gagne, M.; Flynn, B.J.; Andrew, S.F.; Flebbe, D.R.; Mychalowych, A.; Lamb, E.; Davis-Gardner, M.E.; Burnett, M.R.; Serebryannyy, L.A.; Lin, B.C.; et al. Mucosal Adenoviral-vectored Vaccine Boosting Durably Prevents XBB.1.16 Infection in Nonhuman Primates. bioRxiv 2023. [Google Scholar] [CrossRef]
- Darling, T.L.; Harastani, H.H.; Joshi, A.; Bricker, T.L.; Soudani, N.; Seehra, K.; Hassan, A.O.; Diamond, M.S.; Boon, A.C.M. Mucosal immunization with ChAd-SARS-CoV-2-S prevents sequential transmission of SARS-CoV-2 to unvaccinated hamsters. Sci. Adv. 2024, 10, eadp1290. [Google Scholar] [CrossRef]
- Braun, M.R.; Martinez, C.I.; Dora, E.G.; Showalter, L.J.; Mercedes, A.R.; Tucker, S.N. Mucosal immunization with Ad5-based vaccines protects Syrian hamsters from challenge with omicron and delta variants of SARS-CoV-2. Front. Immunol. 2023, 14, 1086035. [Google Scholar] [CrossRef]
- Laghlali, G.; Wiest, M.J.; Karadag, D.; Warang, P.; O’Konek, J.J.; Chang, L.A.; Park, S.; Farazuddin, M.; Landers, J.J.; Janczak, K.W.; et al. Enhanced mucosal B- and T-cell responses against SARS-CoV-2 after heterologous intramuscular mRNA prime/intranasal protein boost vaccination with a combination adjuvant. bioRxiv 2024. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, C.; Yin, L.; Sun, J.; Wang, W.; Li, H.; Zhang, Z.; Chen, S.; Liu, B.; Liu, Z.; et al. Intranasal booster using an Omicron vaccine confers broad mucosal and systemic immunity against SARS-CoV-2 variants. Signal Transduct. Target. Ther. 2023, 8, 167. [Google Scholar] [CrossRef]
- Colwill, K.; Galipeau, Y.; Stuible, M.; Gervais, C.; Arnold, C.; Rathod, B.; Abe, K.T.; Wang, J.H.; Pasculescu, A.; Maltseva, M.; et al. A scalable serology solution for profiling humoral immune responses to SARS-CoV-2 infection and vaccination. Clin. Transl. Immunol. 2022, 11, e1380. [Google Scholar] [CrossRef]
- Lederer, K.; Castano, D.; Gomez Atria, D.; Oguin, T.H., 3rd; Wang, S.; Manzoni, T.B.; Muramatsu, H.; Hogan, M.J.; Amanat, F.; Cherubin, P.; et al. SARS-CoV-2 mRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated with Neutralizing Antibody Generation. Immunity 2020, 53, 1281–1295.e1285. [Google Scholar] [CrossRef]
- Khoury, D.S.; Schlub, T.E.; Cromer, D.; Steain, M.; Fong, Y.; Gilbert, P.B.; Subbarao, K.; Triccas, J.A.; Kent, S.J.; Davenport, M.P. Correlates of Protection, Thresholds of Protection, and Immunobridging among Persons with SARS-CoV-2 Infection. Emerg. Infect. Dis. 2023, 29, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef]
- Walls, A.C.; Miranda, M.C.; Schafer, A.; Pham, M.N.; Greaney, A.; Arunachalam, P.S.; Navarro, M.J.; Tortorici, M.A.; Rogers, K.; O’Connor, M.A.; et al. Elicitation of broadly protective sarbecovirus immunity by receptor-binding domain nanoparticle vaccines. Cell 2021, 184, 5432–5447.e5416. [Google Scholar] [CrossRef]
- Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Viant, C.; Gaebler, C.; Cipolla, M.; Hoffmann, H.H.; Oliveira, T.Y.; Oren, D.A.; et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci. Transl. Med. 2021, 13, eabf1555. [Google Scholar] [CrossRef]
- Neurath, M.F.; Finotto, S.; Glimcher, L.H. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 2002, 8, 567–573. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Faustini, S.E.; Jossi, S.E.; Perez-Toledo, M.; Shields, A.M.; Allen, J.D.; Watanabe, Y.; Newby, M.L.; Cook, A.; Willcox, C.R.; Salim, M.; et al. Development of a high-sensitivity ELISA detecting IgG, IgA and IgM antibodies to the SARS-CoV-2 spike glycoprotein in serum and saliva. Immunology 2021, 164, 135–147. [Google Scholar] [CrossRef]
- Isho, B.; Abe, K.T.; Zuo, M.; Jamal, A.J.; Rathod, B.; Wang, J.H.; Li, Z.; Chao, G.; Rojas, O.L.; Bang, Y.M.; et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci. Immunol. 2020, 5, eabe5511. [Google Scholar] [CrossRef]
- Pisanic, N.; Randad, P.R.; Kruczynski, K.; Manabe, Y.C.; Thomas, D.L.; Pekosz, A.; Klein, S.L.; Betenbaugh, M.J.; Clarke, W.A.; Laeyendecker, O.; et al. COVID-19 Serology at Population Scale: SARS-CoV-2-Specific Antibody Responses in Saliva. J. Clin. Microbiol. 2020, 59, 1–13. [Google Scholar] [CrossRef]
- Reynolds, H.Y. Immunoglobulin G and its function in the human respiratory tract. Mayo Clin. Proc. 1988, 63, 161–174. [Google Scholar] [CrossRef]
- Wu, Y.; Kang, L.; Guo, Z.; Liu, J.; Liu, M.; Liang, W. Incubation Period of COVID-19 Caused by Unique SARS-CoV-2 Strains: A Systematic Review and Meta-analysis. JAMA Netw. Open 2022, 5, e2228008. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.C.; Ng, K.C.; Ching, R.H.H.; Lai, K.L.; Kam, T.T.; Gu, H.; Sit, K.Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Perez, P.; Kato, T.; Mikami, Y.; Okuda, K.; Gilmore, R.C.; Conde, C.D.; Gasmi, B.; Stein, S.; Beach, M.; et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 2021, 27, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. Exploiting Mucosal Immunity for Antiviral Vaccines. Annu. Rev. Immunol. 2016, 34, 575–608. [Google Scholar] [CrossRef]
- Suzuki, T.; Kawaguchi, A.; Ainai, A.; Tamura, S.; Ito, R.; Multihartina, P.; Setiawaty, V.; Pangesti, K.N.; Odagiri, T.; Tashiro, M.; et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc. Natl. Acad. Sci. USA 2015, 112, 7809–7814. [Google Scholar] [CrossRef]
- Ramirez, S.I.; Faraji, F.; Hills, L.B.; Lopez, P.G.; Goodwin, B.; Stacey, H.D.; Sutton, H.J.; Hastie, K.M.; Saphire, E.O.; Kim, H.J.; et al. Immunological memory diversity in the human upper airway. Nature 2024, 632, 630–636. [Google Scholar] [CrossRef]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef]
- Corthesy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef]
- Renegar, K.B.; Small, P.A., Jr.; Boykins, L.G.; Wright, P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004, 173, 1978–1986. [Google Scholar] [CrossRef]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2006, 208, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.A. The structure and function of human IgA. Biochem. J. 1990, 271, 285–296. [Google Scholar] [CrossRef]
- Isho, B.; Florescu, A.; Wang, A.A.; Gommerman, J.L. Fantastic IgA plasma cells and where to find them. Immunol. Rev. 2021, 303, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Rojas, O.L.; Probstel, A.K.; Porfilio, E.A.; Wang, A.A.; Charabati, M.; Sun, T.; Lee, D.S.W.; Galicia, G.; Ramaglia, V.; Ward, L.A.; et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 2019, 177, 492–493. [Google Scholar] [CrossRef]
- Nahass, G.R.; Salomon-Shulman, R.E.; Blacker, G.; Haider, K.; Brotherton, R.; Teague, K.; Yiu, Y.Y.; Brewer, R.E.; Galloway, S.D.; Hansen, P. Intramuscular SARS-CoV-2 vaccines elicit varying degrees of plasma and salivary antibody responses as compared to natural infection. Medrxiv 2021. [Google Scholar] [CrossRef]
- Park, S.K.; Lee, C.W.; Park, D.I.; Woo, H.Y.; Cheong, H.S.; Shin, H.C.; Ahn, K.; Kwon, M.J.; Joo, E.J. Detection of SARS-CoV-2 in Fecal Samples from Patients with Asymptomatic and Mild COVID-19 in Korea. Clin. Gastroenterol. Hepatol. 2021, 19, 1387–1394.e1382. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Morone, G.; Palomba, A.; Iosa, M.; Caporaso, T.; De Angelis, D.; Venturiero, V.; Savo, A.; Coiro, P.; Carbone, D.; Gimigliano, F.; et al. Incidence and Persistence of Viral Shedding in COVID-19 Post-acute Patients with Negativized Pharyngeal Swab: A Systematic Review. Front. Med. 2020, 7, 562. [Google Scholar] [CrossRef]
- Schiepers, A.; van’t Wout, M.F.L.; Greaney, A.J.; Zang, T.; Muramatsu, H.; Lin, P.J.C.; Tam, Y.K.; Mesin, L.; Starr, T.N.; Bieniasz, P.D.; et al. Molecular fate-mapping of serum antibody responses to repeat immunization. Nature 2023, 615, 482–489. [Google Scholar] [CrossRef]
- Alsoussi, W.B.; Malladi, S.K.; Zhou, J.Q.; Liu, Z.; Ying, B.; Kim, W.; Schmitz, A.J.; Lei, T.; Horvath, S.C.; Sturtz, A.J.; et al. SARS-CoV-2 Omicron boosting induces de novo B cell response in humans. Nature 2023, 617, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Yisimayi, A.; Song, W.; Wang, J.; Jian, F.; Yu, Y.; Chen, X.; Xu, Y.; Yang, S.; Niu, X.; Xiao, T.; et al. Repeated Omicron exposures override ancestral SARS-CoV-2 immune imprinting. Nature 2024, 625, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Davis, W.G.; Sambhara, S.; Jacob, J. Strategies to alleviate original antigenic sin responses to influenza viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 13751–13756. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Sidney, J.; Vita, R.; Peters, B.; Crotty, S.; Weiskopf, D.; Sette, A. SARS-CoV-2 human T cell epitopes: Adaptive immune response against COVID-19. Cell Host Microbe 2022, 30, 1788. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Administration (Prime–Boost) * | Type of Vaccine Antigenic Formulation | Prime (Day 1) | Boost (Day 21) |
|---|---|---|---|
| IM–IN | - | PBS | PBS |
| IM–IN | - | Monovalent mRNA-LNP | PBS |
| IM–IN | - | Bivalent mRNA-LNP | PBS |
| IN–IN | Homologous | Wu-1 protein | Wu-1 protein |
| IM–IM | Homologous | Monovalent mRNA-LNP | Monovalent mRNA-LNP |
| IM–IN | Homologous | Monovalent mRNA-LNP | Wu-1 protein |
| IM–IN | Heterologous | Monovalent mRNA-LNP | BA4/5 protein |
| IM–IN | Heterologous | Monovalent mRNA-LNP | Wu-1 + BA4/5 protein |
| IM–IM | Homologous | Bivalent mRNA-LNP | Bivalent mRNA-LNP |
| IM–IN | Homologous | Bivalent mRNA-LNP | Wu-1 protein |
| IM–IN | Homologous | Bivalent mRNA-LNP | BA4/5 protein |
| IM–IN | Homologous | Bivalent mRNA-LNP | Wu-1 + BA4/5 protein |
| IM–IN | Heterologous | Bivalent mRNA-LNP | NL63 protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maltseva, M.; Galipeau, Y.; McCluskie, P.; Castonguay, N.; Cooper, C.L.; Langlois, M.-A. Systemic and Mucosal Antibody Responses to SARS-CoV-2 Variant-Specific Prime-and-Boost and Prime-and-Spike Vaccination: A Comparison of Intramuscular and Intranasal Bivalent Vaccine Administration in a Murine Model. Vaccines 2025, 13, 351. https://doi.org/10.3390/vaccines13040351
Maltseva M, Galipeau Y, McCluskie P, Castonguay N, Cooper CL, Langlois M-A. Systemic and Mucosal Antibody Responses to SARS-CoV-2 Variant-Specific Prime-and-Boost and Prime-and-Spike Vaccination: A Comparison of Intramuscular and Intranasal Bivalent Vaccine Administration in a Murine Model. Vaccines. 2025; 13(4):351. https://doi.org/10.3390/vaccines13040351
Chicago/Turabian StyleMaltseva, Mariam, Yannick Galipeau, Pauline McCluskie, Nicolas Castonguay, Curtis L. Cooper, and Marc-André Langlois. 2025. "Systemic and Mucosal Antibody Responses to SARS-CoV-2 Variant-Specific Prime-and-Boost and Prime-and-Spike Vaccination: A Comparison of Intramuscular and Intranasal Bivalent Vaccine Administration in a Murine Model" Vaccines 13, no. 4: 351. https://doi.org/10.3390/vaccines13040351
APA StyleMaltseva, M., Galipeau, Y., McCluskie, P., Castonguay, N., Cooper, C. L., & Langlois, M.-A. (2025). Systemic and Mucosal Antibody Responses to SARS-CoV-2 Variant-Specific Prime-and-Boost and Prime-and-Spike Vaccination: A Comparison of Intramuscular and Intranasal Bivalent Vaccine Administration in a Murine Model. Vaccines, 13(4), 351. https://doi.org/10.3390/vaccines13040351