
Multi-Antigen Viral-Vectored Vaccine Protects Against SARS-CoV-2 and Variants in a Lethal hACE2 Transgenic Mouse Model

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vaccine Construction and Characterization
2.2. In Vivo Mouse Vaccination and Infection Experiments
2.3. Plaque Reduction Neutralization Test (PRNT)
2.4. Binding Antibody
2.5. T Cell Analysis
2.6. Viral Load Analysis
2.7. Immunohistochemistry
2.8. Luminex
2.9. Statistical Analysis
3. Results
3.1. Design and Characterization of Multi-Antigen MVA-VLP Vaccine Candidate
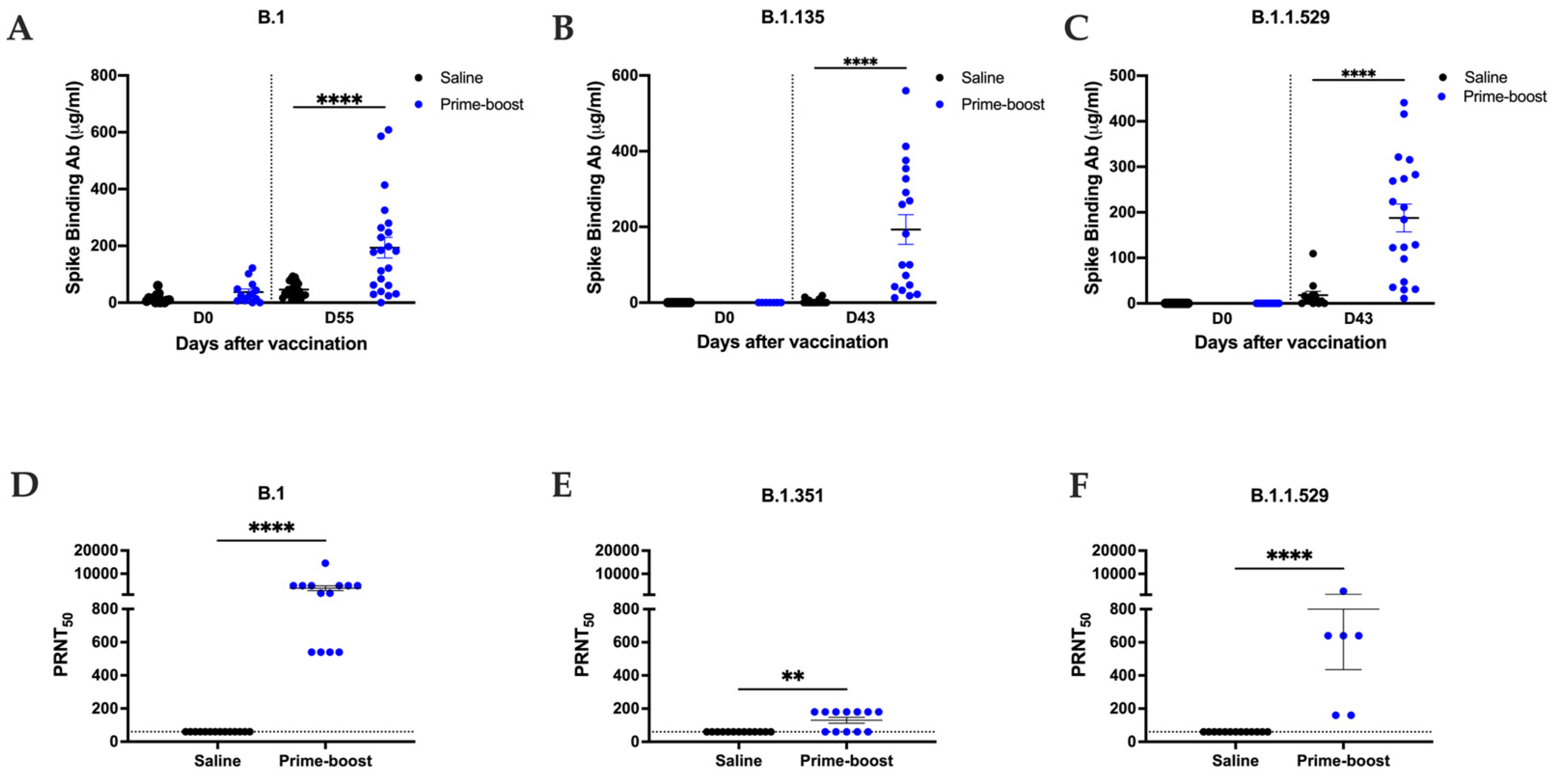
3.2. GEO-CM02 Vaccination Elicits Neutralizing Antibodies and Functional T Cell Responses in hACE2 Mice
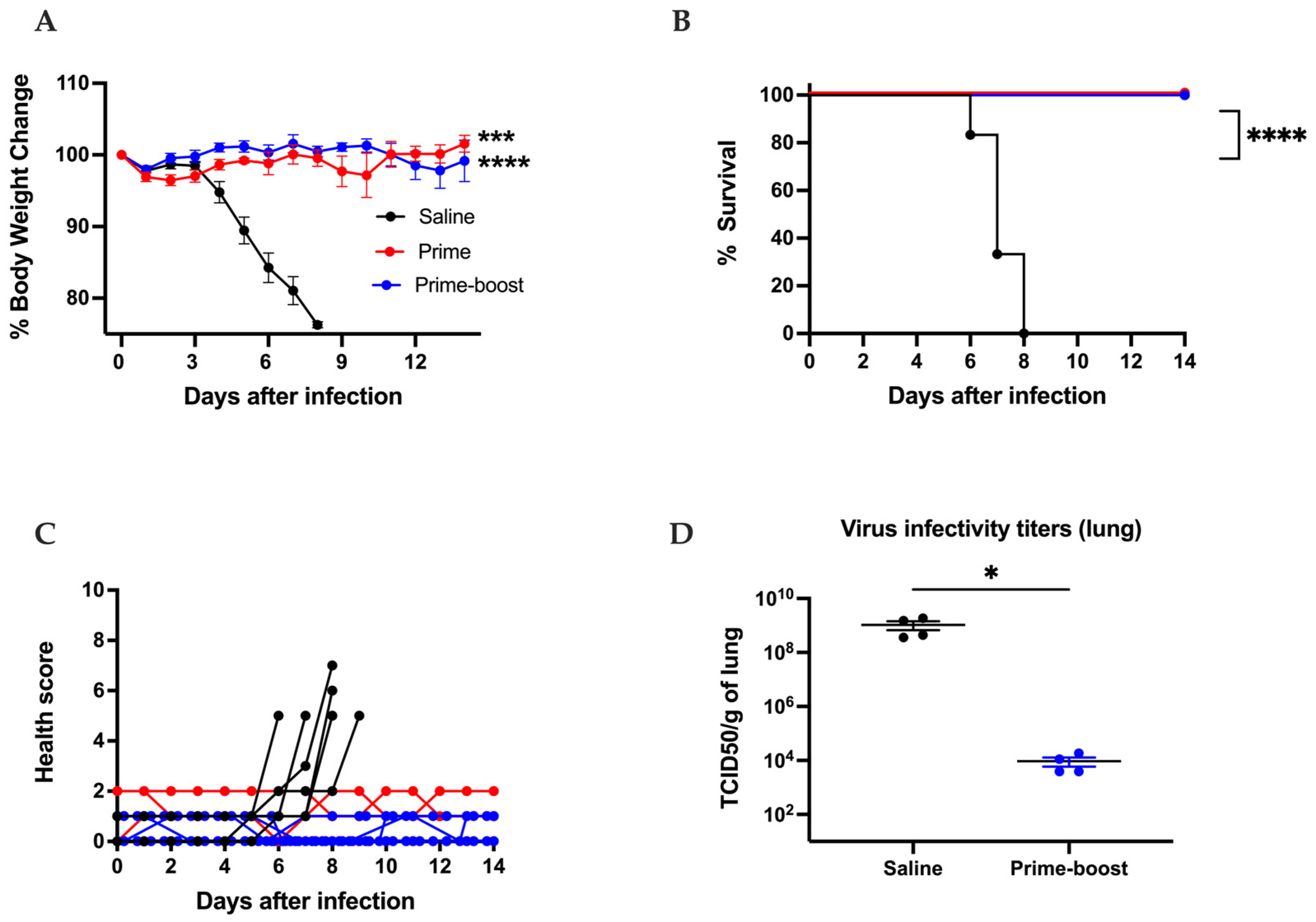
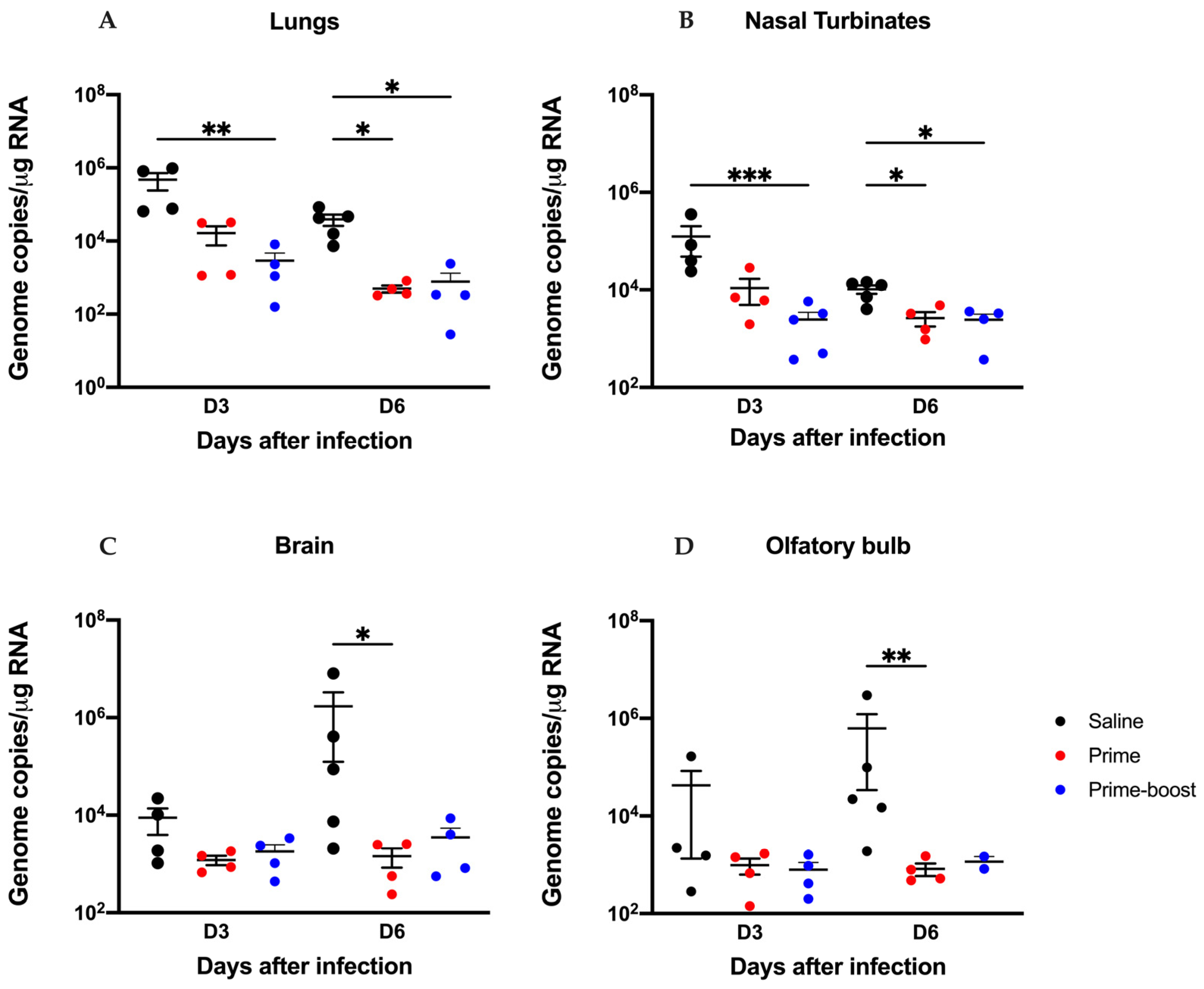
3.3. The GEO-CM02 Vaccine Provides Protection Against Lethal SARS-CoV-2 Variants in K18-hACE2 Mice
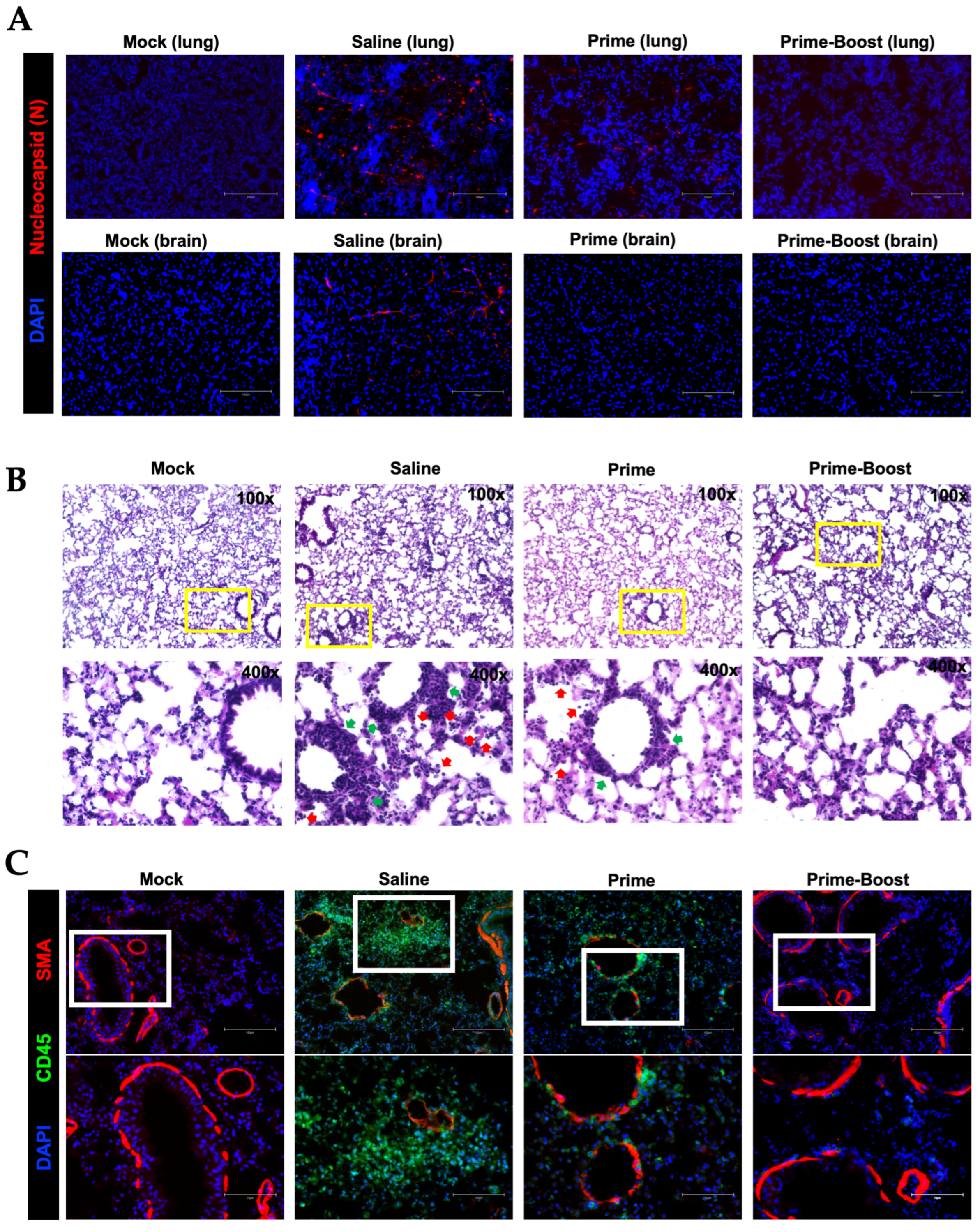
3.4. Dramatic Reduction in Lung Pathology and Inflammatory Markers in GEO-CM02-Vaccinated Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef] [PubMed]
- FDA. COVID-19 Vaccines for 2023–2024. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines-2023-2024 (accessed on 1 January 2025).
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; Pajon, R.; Smith, G.; Glenn, G.M.; Shi, W.; Korber, B.; Montefiori, D.C. Neutralization of SARS-CoV-2 variants B. 1.429 and B. 1.351. N. Engl. J. Med. 2021, 384, 2352–2354. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Hachmann, N.P.; Miller, J.; Collier, A.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K. Neutralization escape by SARS-CoV-2 Omicron subvariants BA. 2.12. 1, BA. 4, and BA. 5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X. BA. 2.12. 1, BA. 4 and BA. 5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Yang, J.; Han, M.; Wang, L.; Wang, L.; Xu, T.; Wu, L.; Ma, J.; Wong, G.; Liu, W.; Gao, G.F. Relatively rapid evolution rates of SARS-CoV-2 spike gene at the primary stage of massive vaccination. Biosaf. Health 2022, 4, 228–233. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, D.-U.; Noh, J.Y.; Kim, S.; Park, S.-H.; Jeong, H.W.; Shin, E.-C. T cell epitopes in SARS-CoV-2 proteins are substantially conserved in the Omicron variant. Cell. Mol. Immunol. 2022, 19, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Primorac, D.; Brlek, P.; Matišić, V.; Molnar, V.; Vrdoljak, K.; Zadro, R.; Parčina, M. Cellular immunity—The key to long-term protection in individuals recovered from SARS-CoV-2 and after vaccination. Vaccines 2022, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Le Bert, N.; Qui, M.; Tan, A.T. SARS-CoV-2-specific T cells in infection and vaccination. Cell. Mol. Immunol. 2021, 18, 2307–2312. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Oja, A.E.; Saris, A.; Ghandour, C.A.; Kragten, N.A.; Hogema, B.M.; Nossent, E.J.; Heunks, L.M.; Cuvalay, S.; Slot, E.; Linty, F. Divergent SARS-CoV-2-specific T-and B-cell responses in severe but not mild COVID-19 patients. Eur. J. Immunol. 2020, 50, 1998–2012. [Google Scholar] [CrossRef]
- Moderbacher, C.R.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 2020, 183, 996–1012.E19. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 2020, 183, 158–168.E14. [Google Scholar] [CrossRef]
- Tan, Y.; Liu, F.; Xu, X.; Ling, Y.; Huang, W.; Zhu, Z.; Guo, M.; Lin, Y.; Fu, Z.; Liang, D. Durability of neutralizing antibodies and T-cell response post SARS-CoV-2 infection. Front. Med. 2020, 14, 746–751. [Google Scholar] [CrossRef]
- Ni, L.; Ye, F.; Cheng, M.-L.; Feng, Y.; Deng, Y.-Q.; Zhao, H.; Wei, P.; Ge, J.; Gou, M.; Li, X. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 2020, 52, 971–977.E3. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J.; Beale, R. Immunity to SARS-CoV-2 variants of concern. Science 2021, 371, 1103–1104. [Google Scholar] [CrossRef]
- Chiuppesi, F.; Nguyen, V.H.; Park, Y.; Contreras, H.; Karpinski, V.; Faircloth, K.; Nguyen, J.; Kha, M.; Johnson, D.; Martinez, J. Synthetic multiantigen MVA vaccine COH04S1 protects against SARS-CoV-2 in Syrian hamsters and non-human primates. npj Vaccines 2022, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.; Chia, A.; Smith, G.J. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020, 181, 1489–1501.E15. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 virus-like particles by mammalian expression system. Front. Bioeng. Biotechnol. 2020, 8, 862. [Google Scholar]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Liu, L.; Chavan, R.; Feinberg, M.B. Dendritic cells are preferentially targeted among hematolymphocytes by Modified Vaccinia Virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol. 2008, 9, 15. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Drexler, I.; Ludwig, H.; Erfle, V.; Peschel, C.; Bernhard, H.; Sutter, G. Infection of human dendritic cells with recombinant vaccinia virus MVA reveals general persistence of viral early transcription but distinct maturation-dependent cytopathogenicity. Virology 2006, 350, 276–288. [Google Scholar] [CrossRef]
- Altenburg, A.F.; van de Sandt, C.E.; Li, B.W.; MacLoughlin, R.J.; Fouchier, R.A.; van Amerongen, G.; Volz, A.; Hendriks, R.W.; de Swart, R.L.; Sutter, G. Modified vaccinia virus Ankara preferentially targets antigen presenting cells in vitro, ex vivo and in vivo. Sci. Rep. 2017, 7, 8580. [Google Scholar] [CrossRef]
- Gasteiger, G.; Kastenmuller, W.; Ljapoci, R.; Sutter, G.; Drexler, I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J. Virol. 2007, 81, 11925–11936. [Google Scholar] [CrossRef]
- Shen, X.; Wong, S.; Buck, C.B.; Zhang, J.; Siliciano, R.F. Direct priming and cross-priming contribute differentially to the induction of CD8+ CTL following exposure to vaccinia virus via different routes. J. Immunol. 2002, 169, 4222–4229. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Elizaga, M.L.; Sato, A.; Qin, L.; Cardinali, M.; Hay, C.M.; Hural, J.; DeRosa, S.C.; DeFawe, O.D.; Tomaras, G.D. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J. Infect. Dis. 2011, 203, 610–619. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Elizaga, M.L.; Seaton, K.; Tomaras, G.D.; Montefiori, D.C.; Sato, A.; Hural, J.; DeRosa, S.C.; Kalams, S.A.; McElrath, M.J. Specificity and 6-month durability of immune responses induced by DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J. Infect. Dis. 2014, 210, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Salvato, M.S.; Domi, A.; Guzmán-Cardozo, C.; Medina-Moreno, S.; Zapata, J.C.; Hsu, H.; McCurley, N.; Basu, R.; Hauser, M.; Hellerstein, M. A single dose of modified vaccinia Ankara expressing Lassa virus-like particles protects mice from lethal intra-cerebral virus challenge. Pathogens 2019, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Domi, A.; Feldmann, F.; Basu, R.; McCurley, N.; Shifflett, K.; Emanuel, J.; Hellerstein, M.S.; Guirakhoo, F.; Orlandi, C.; Flinko, R. A single dose of modified vaccinia Ankara expressing Ebola virus like particles protects nonhuman primates from lethal Ebola virus challenge. Sci. Rep. 2018, 8, 864. [Google Scholar] [CrossRef]
- Ramírez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B-and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef]
- Ramírez, J.C.; Gherardi, M.M.; Rodríguez, D.; Esteban, M. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. J. Virol. 2000, 74, 7651–7655. [Google Scholar] [CrossRef]
- Brault, A.C.; Domi, A.; McDonald, E.M.; Talmi-Frank, D.; McCurley, N.; Basu, R.; Robinson, H.L.; Hellerstein, M.; Duggal, N.K.; Bowen, R.A. A Zika vaccine targeting NS1 protein protects immunocompetent adult mice in a lethal challenge model. Sci. Rep. 2017, 7, 14769. [Google Scholar] [CrossRef]
- Malherbe, D.C.; Domi, A.; Hauser, M.J.; Meyer, M.; Gunn, B.M.; Alter, G.; Bukreyev, A.; Guirakhoo, F. Modified vaccinia Ankara vaccine expressing Marburg virus-like particles protects guinea pigs from lethal Marburg virus infection. npj Vaccines 2020, 5, 78. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef]
- Kumar, M.; O’Connell, M.; Namekar, M.; Nerurkar, V.R. Infection with non-lethal West Nile virus Eg101 strain induces immunity that protects mice against the lethal West Nile virus NY99 strain. Viruses 2014, 6, 2328–2339. [Google Scholar] [CrossRef]
- Elsharkawy, A.; Stone, S.; Guglani, A.; Patterson, L.D.; Ge, C.; Dim, C.; Miano, J.M.; Kumar, M. Omicron XBB.1.5 subvariant causes severe pulmonary disease in K18-hACE-2 mice. Front. Microbiol. 2024, 15, 1466980. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.; Rothan, H.A.; Natekar, J.P.; Kumari, P.; Sharma, S.; Pathak, H.; Arora, K.; Auroni, T.T.; Kumar, M. SARS-CoV-2 variants of concern infect the respiratory tract and induce inflammatory response in wild-type laboratory mice. Viruses 2021, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Stone, S.; Natekar, J.; Kumari, P.; Arora, K.; Kumar, M. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-CoV-2) replication and attenuates inflammation in human cells. Virology 2020, 547, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Roe, K.; Orillo, B.; Muruve, D.A.; Nerurkar, V.R.; Gale, M., Jr.; Verma, S. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J. Virol. 2013, 87, 3655–3667. [Google Scholar] [CrossRef]
- Oh, S.-J.; Kumari, P.; Auroni, T.T.; Stone, S.; Pathak, H.; Elsharkawy, A.; Natekar, J.P.; Shin, O.S.; Kumar, M. Upregulation of Neuroinflammation-Associated Genes in the Brain of SARS-CoV-2-Infected Mice. Pathogens 2024, 13, 528. [Google Scholar] [CrossRef]
- Callaway, E. Remember Beta? New data reveal variant’s deadly powers. Nature 2021. [Google Scholar] [CrossRef]
- Soni, M.; Migliori, E.; Fu, J.; Assal, A.; Chan, H.T.; Pan, J.; Khatiwada, P.; Ciubotariu, R.; May, M.S.; Pereira, M.; et al. The prospect of universal coronavirus immunity: A characterization of reciprocal and non-reciprocal T cell responses against SARS-CoV2 and common human coronaviruses. bioRxiv 2023. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, Q.; Gu, X.; Ren, L.; Huang, T.; Li, Y.; Zhang, H.; Liu, Y.; Zhong, J.; Wang, X.; et al. Durability and cross-reactive immune memory to SARS-CoV-2 in individuals 2 years after recovery from COVID-19: A longitudinal cohort study. Lancet Microbe 2024, 5, e24–e33. [Google Scholar] [CrossRef]
- Arieta, C.M.; Xie, Y.J.; Rothenberg, D.A.; Diao, H.; Harjanto, D.; Meda, S.; Marquart, K.; Koenitzer, B.; Sciuto, T.E.; Lobo, A.; et al. The T-cell-directed vaccine BNT162b4 encoding conserved non-spike antigens protects animals from severe SARS-CoV-2 infection. Cell 2023, 186, 2392–2409.E21. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, U.J.; Bukreyev, A.; Yang, L.; Lamirande, E.W.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. USA 2004, 101, 9804–9809. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, Y.; Huang, B.; Han, D.; Wang, W.; Huang, M.; Zhai, C.; Zhao, Z.; Yang, R.; Zhao, Y.; et al. DNA Vaccines Expressing the Envelope and Membrane Proteins Provide Partial Protection Against SARS-CoV-2 in Mice. Front. Immunol. 2022, 13, 827605. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Miura, K.; Suzuki, Y.; Ishida, K.; Arakawa, M.; Wu, H.; Fujioka, Y.; Emi, A.; Maeda, K.; Hamajima, R.; Nakano, T.; et al. Distinct motifs in the E protein are required for SARS-CoV-2 virus particle formation and lysosomal deacidification in host cells. J. Virol. 2023, 97, e0042623. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stone, S.; Elsharkawy, A.; Burleson, J.D.; Hauser, M.; Domi, A.; Kumari, P.; Nabi, Z.; Natekar, J.P.; Porto, M.; Backstedt, B.; et al. Multi-Antigen Viral-Vectored Vaccine Protects Against SARS-CoV-2 and Variants in a Lethal hACE2 Transgenic Mouse Model. Vaccines 2025, 13, 411. https://doi.org/10.3390/vaccines13040411
Stone S, Elsharkawy A, Burleson JD, Hauser M, Domi A, Kumari P, Nabi Z, Natekar JP, Porto M, Backstedt B, et al. Multi-Antigen Viral-Vectored Vaccine Protects Against SARS-CoV-2 and Variants in a Lethal hACE2 Transgenic Mouse Model. Vaccines. 2025; 13(4):411. https://doi.org/10.3390/vaccines13040411
Chicago/Turabian StyleStone, Shannon, Amany Elsharkawy, J. D. Burleson, Mary Hauser, Arban Domi, Pratima Kumari, Zainab Nabi, Janhavi P. Natekar, Maciel Porto, Brian Backstedt, and et al. 2025. "Multi-Antigen Viral-Vectored Vaccine Protects Against SARS-CoV-2 and Variants in a Lethal hACE2 Transgenic Mouse Model" Vaccines 13, no. 4: 411. https://doi.org/10.3390/vaccines13040411
APA StyleStone, S., Elsharkawy, A., Burleson, J. D., Hauser, M., Domi, A., Kumari, P., Nabi, Z., Natekar, J. P., Porto, M., Backstedt, B., Newman, M., Oruganti, S. R., & Kumar, M. (2025). Multi-Antigen Viral-Vectored Vaccine Protects Against SARS-CoV-2 and Variants in a Lethal hACE2 Transgenic Mouse Model. Vaccines, 13(4), 411. https://doi.org/10.3390/vaccines13040411