Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Western Blot
2.3. Immunofluorescence Assay (IFA)
2.4. Animals
2.5. Animal Immunizations
2.6. Animal Challenge Studies
2.7. ELISpot Assay
2.7.1. For HIV Studies
2.7.2. For Influenza Study
2.7.3. For Zika Studies
2.8. Flow Cytometry:
2.9. ELISA
2.10. Statistical Analysis
3. Results
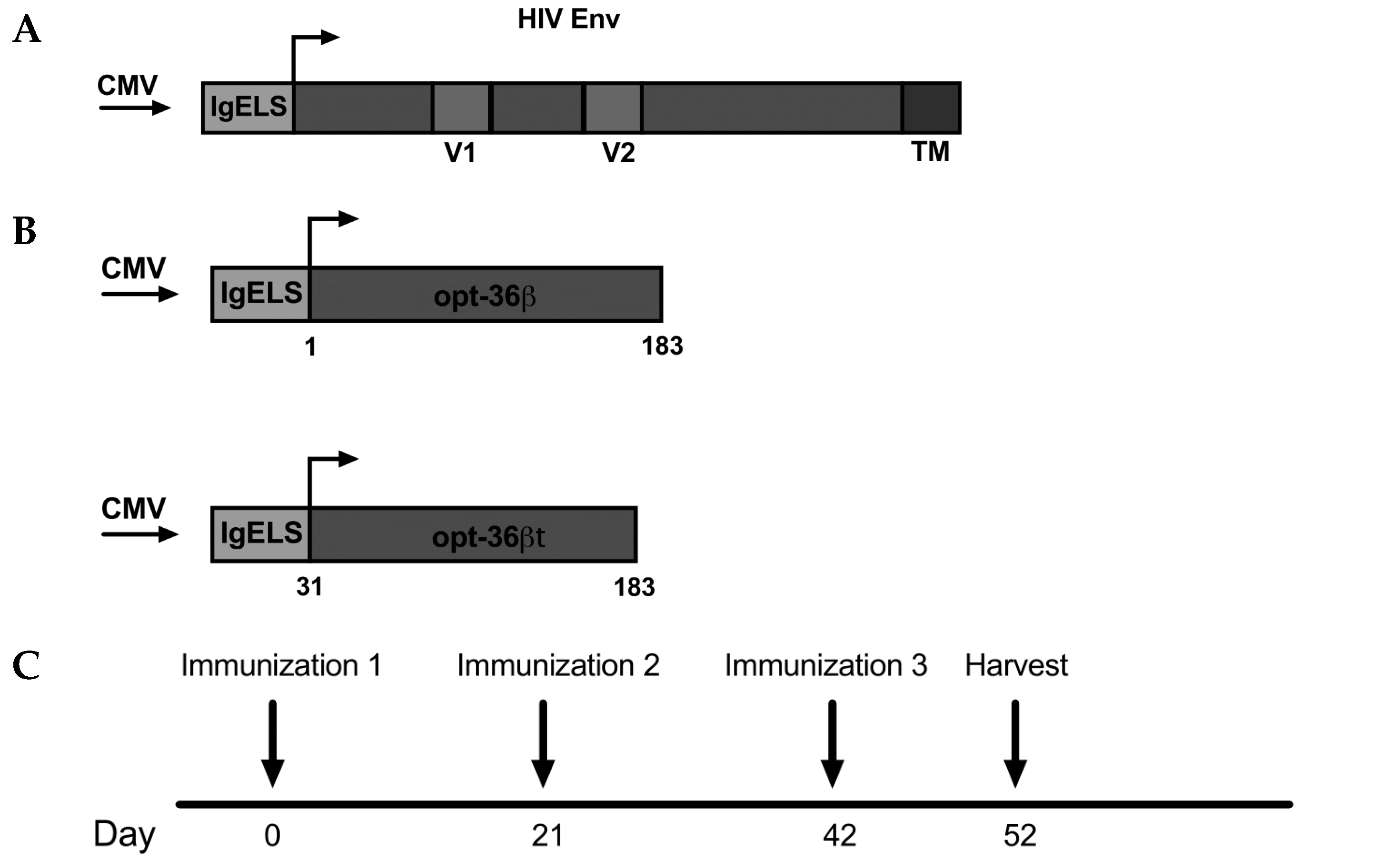
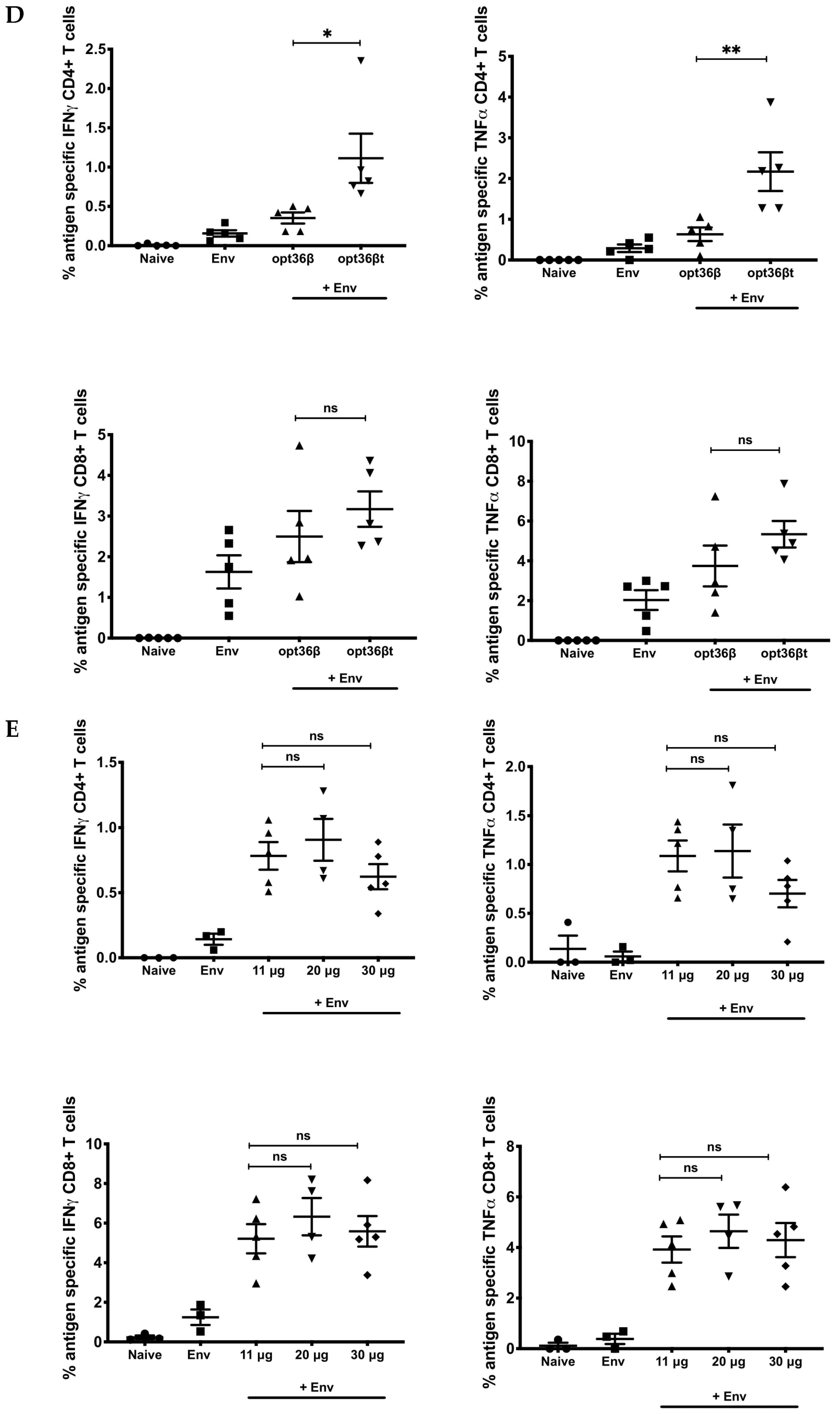
3.1. Opt-36βt Co-Formulation Leads to Enhanced Immune Responses against HIV Env DNA Vaccine Compared to Opt-36β
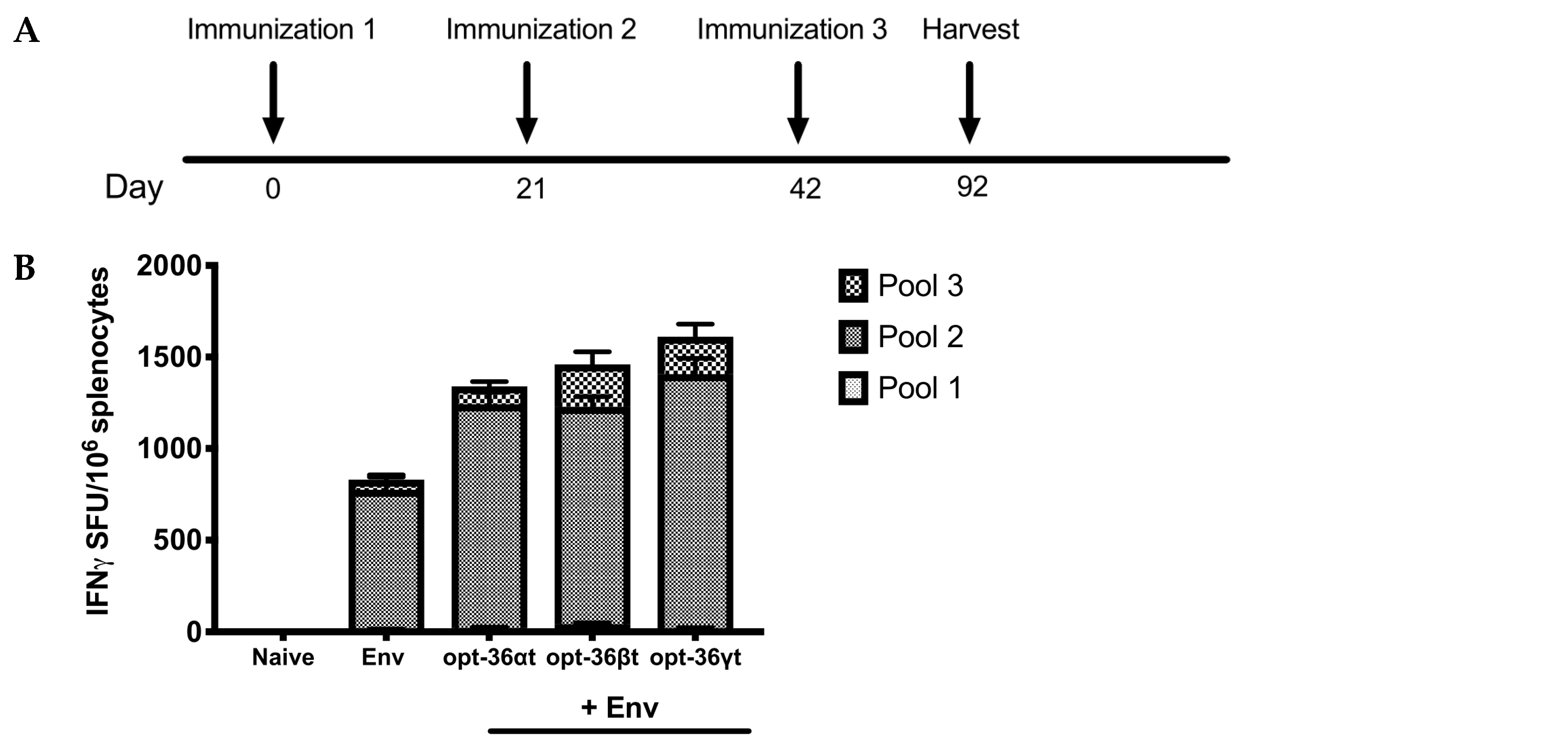
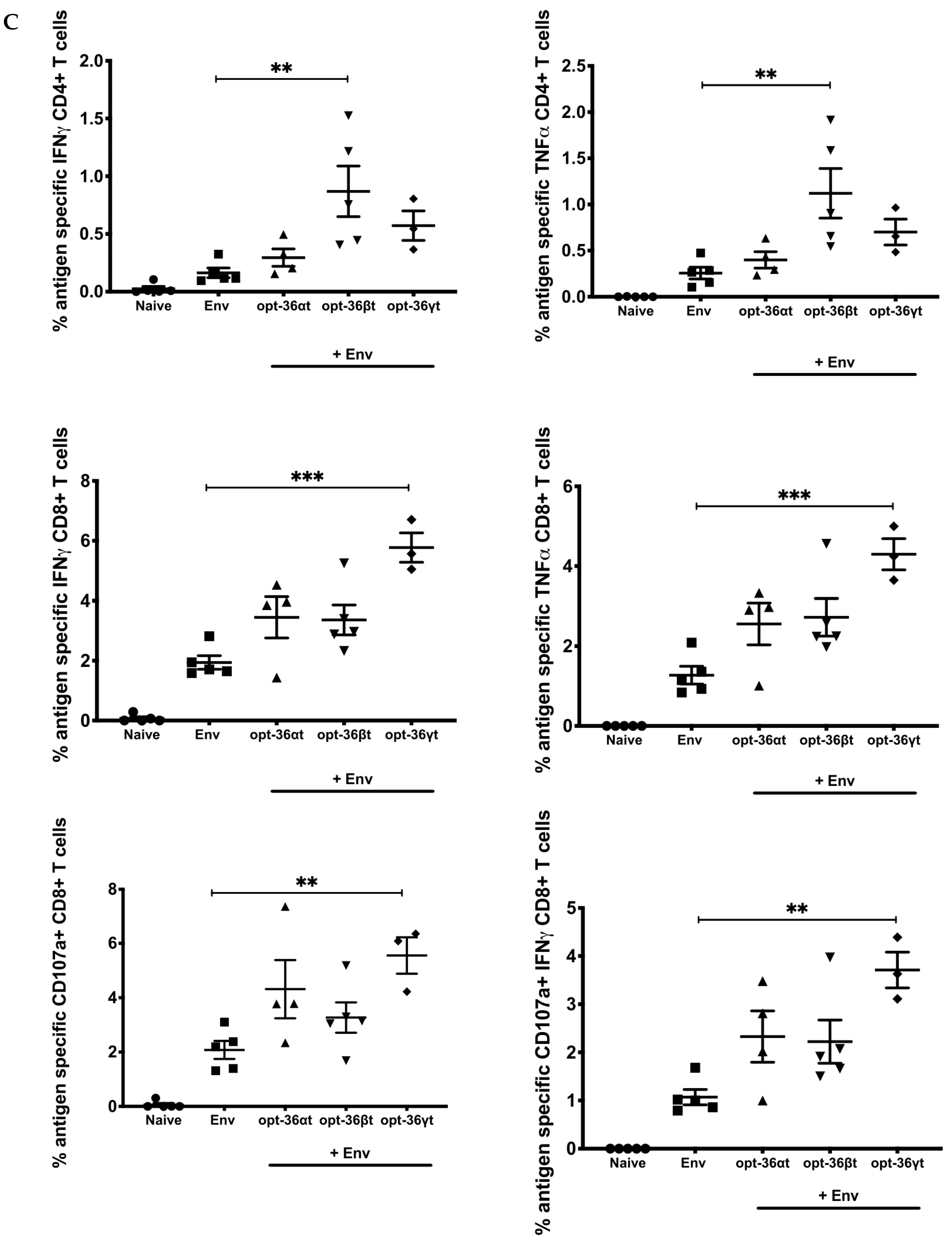
3.2. Opt-36βt and opt-36γt Enhance Immune Responses against HIV Env DNA Vaccine at a Memory Time Point
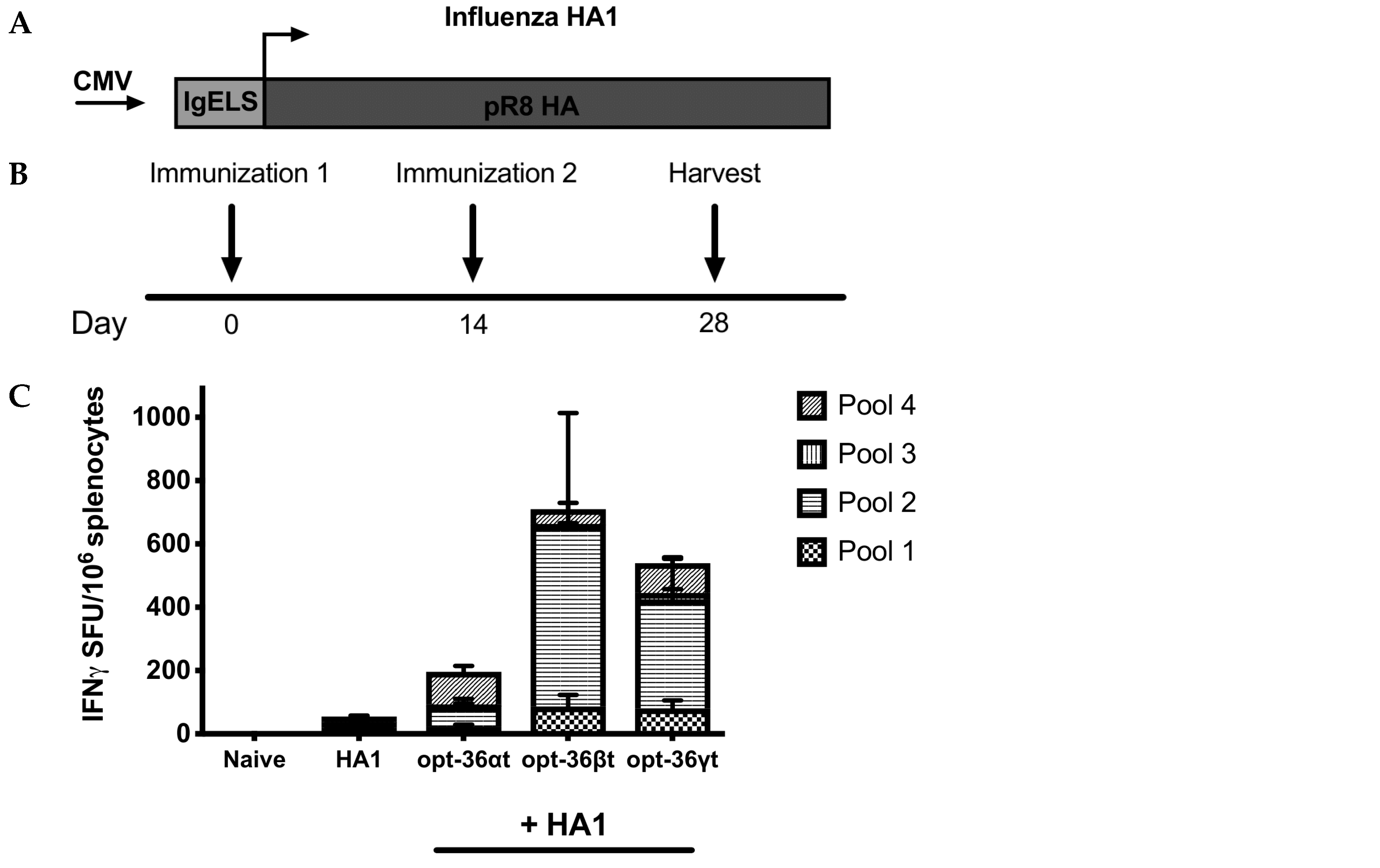
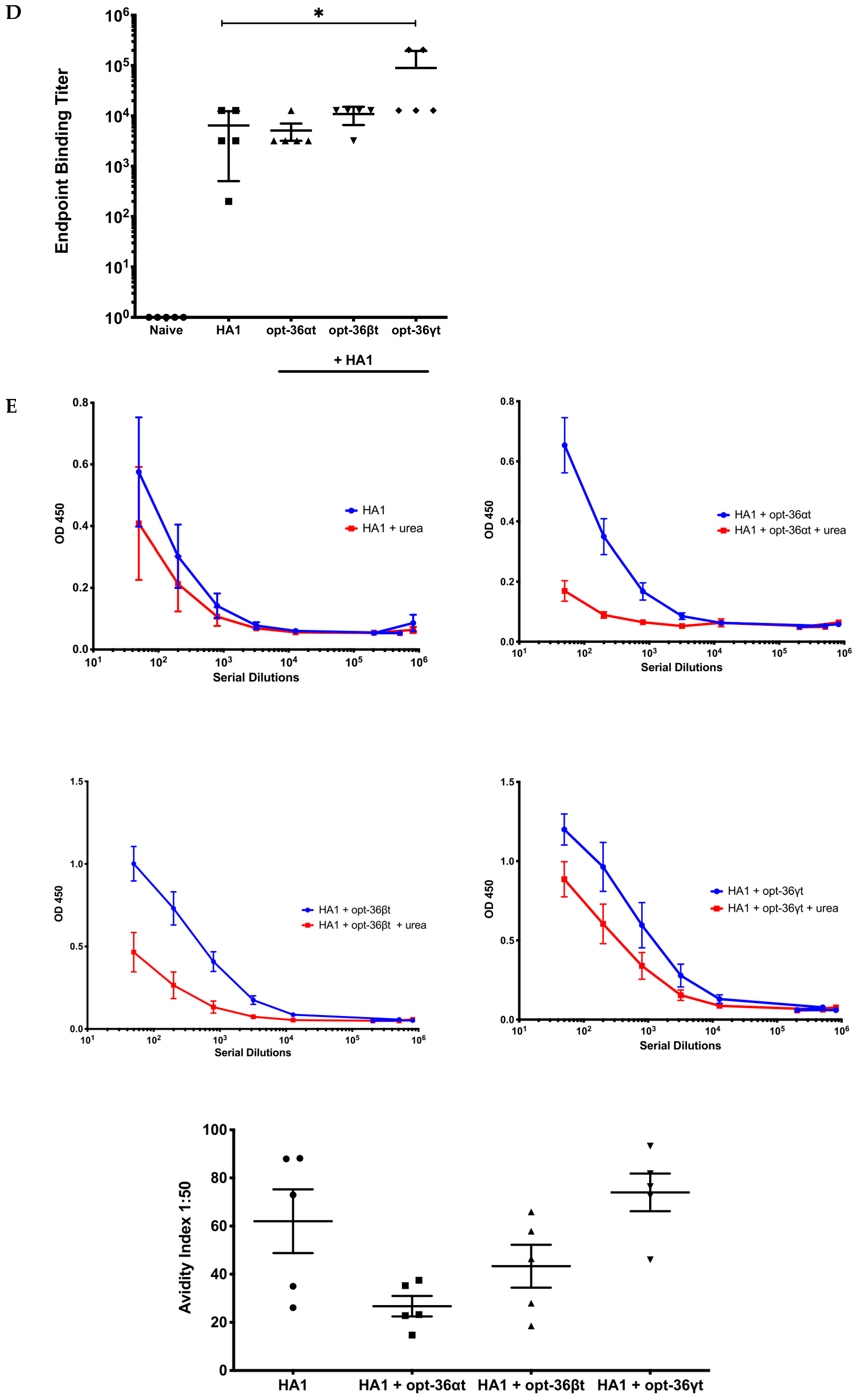
3.3. Opt-36γt Enhances Humoral Immunity in Influenza DNA Vaccine Model
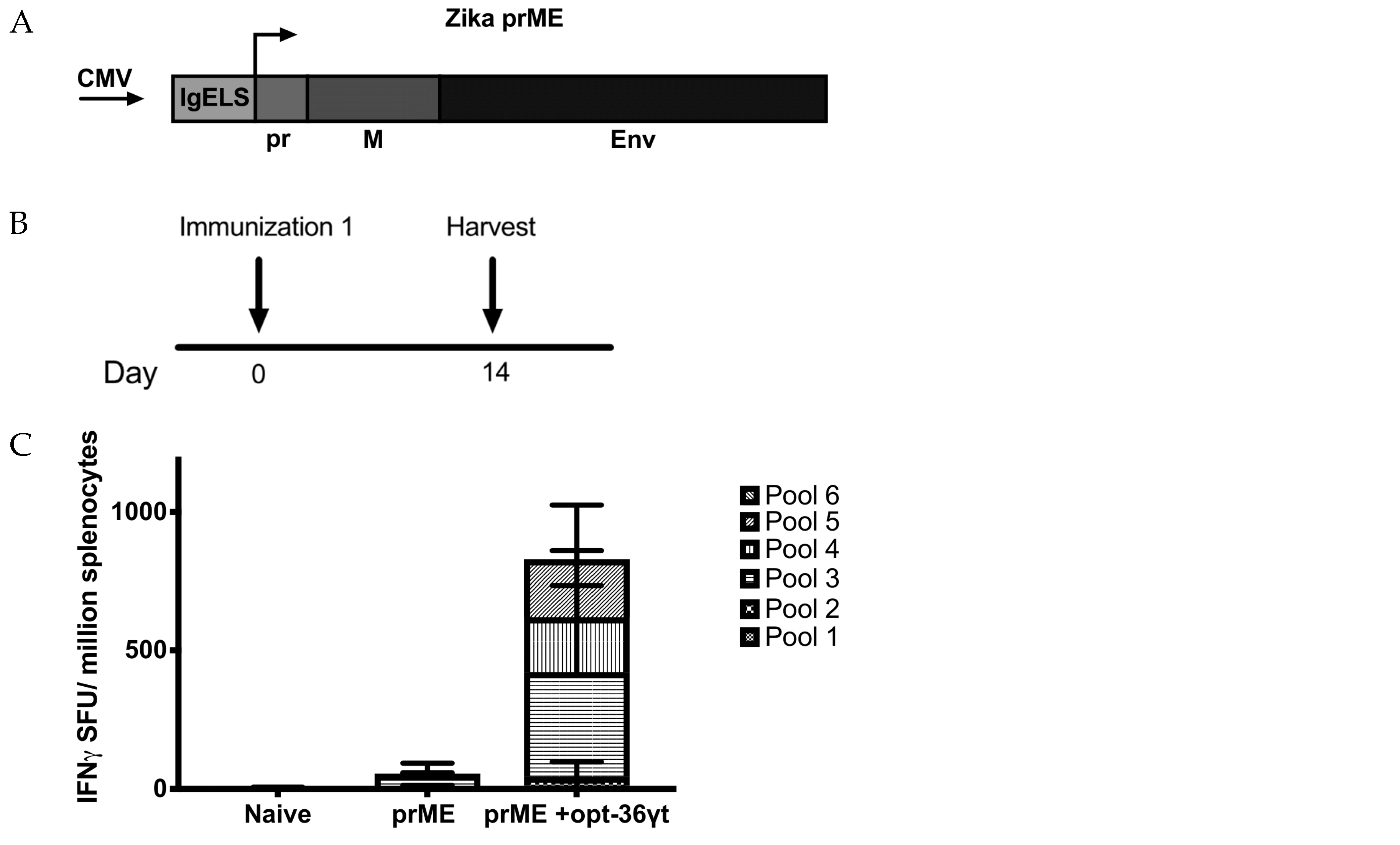
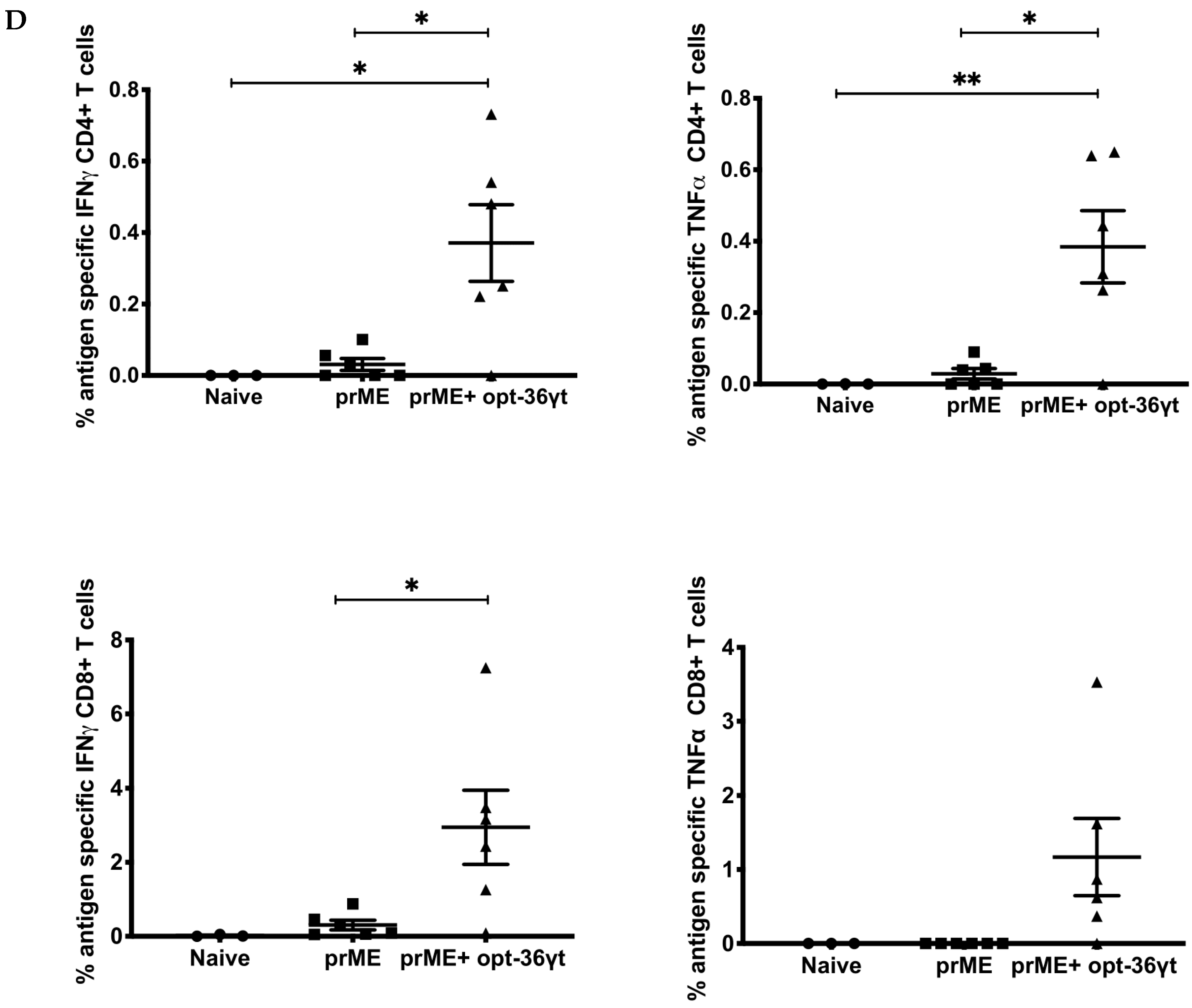
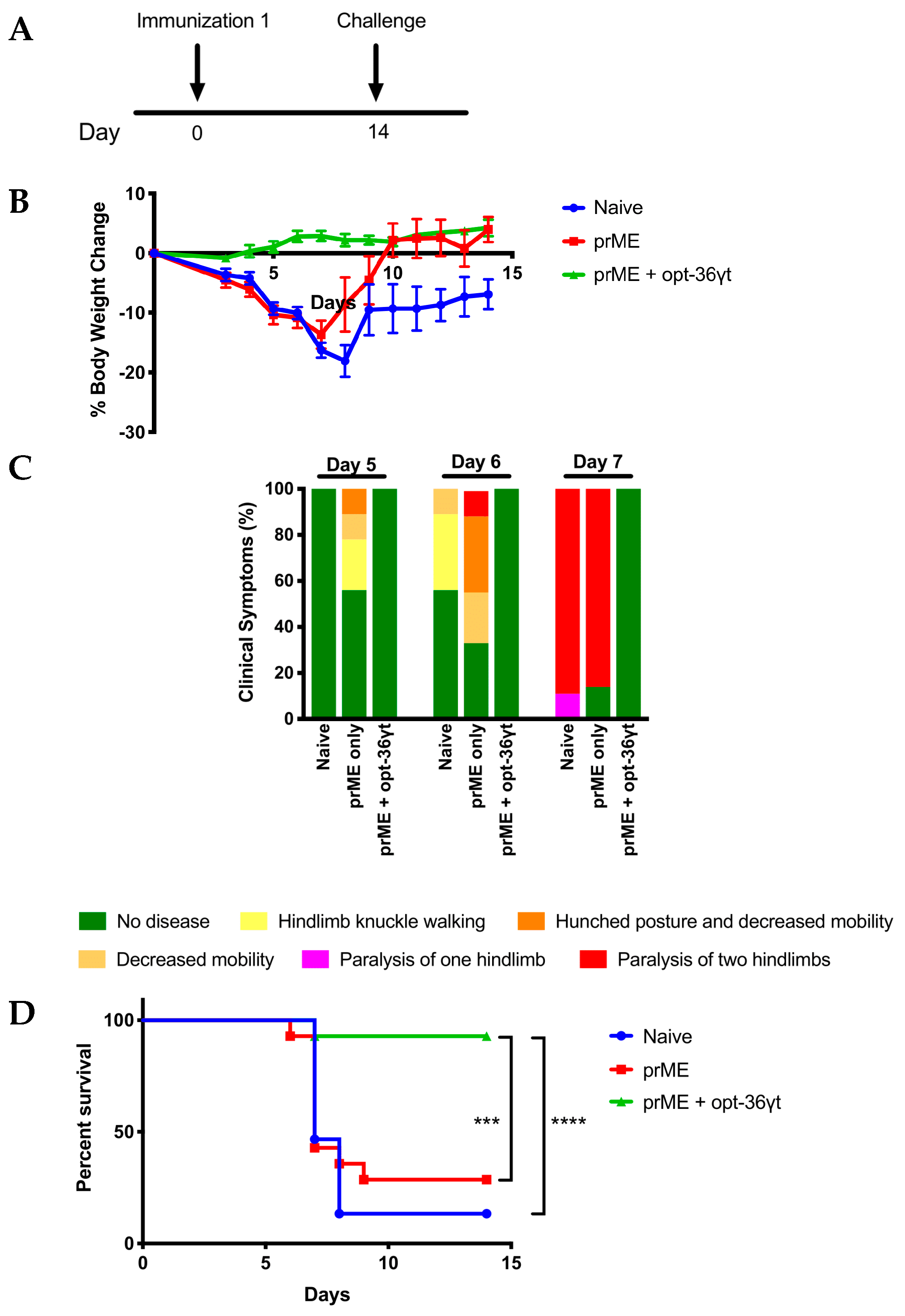
3.4. Opt-36γt Enhances Cellular Immune Responses Induced by a Zika DNA Vaccine Resulting in Enhanced Protection against Zika Challenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lahiri, A.; Das, P. Chakravortty D. Engagement of TLR signaling as adjuvant: Towards smarter vaccine and beyond. Vaccine 2008, 26, 6777–6783. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.S.; Munks, M.W.; Marrack, P. How Do Adjuvants Work? Important Considerations for New Generation Adjuvants. Immunity 2007, 27, 687–690. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Mosca, F.; Tritto, E.; Muzzi, A.; Monaci, E.; Bagnoli, F.; Iavarone, C.; O’Hagan, D.; Rappuoli, R.; De Gregorio, E. Molecular and cellular signatures of human vaccine adjuvants. Proc. Natl. Acad. Sci. USA 2008, 105, 10501–10506. [Google Scholar] [CrossRef] [Green Version]
- Ragupathi, G.; Gardner, J.R.; Livingston, P.O.; Gin, D.Y. Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 2011, 10, 463–470. [Google Scholar] [CrossRef]
- Shah, R.R.; Hassett, K.J.; Brito, L.A. Overview of Vaccine Adjuvants: Introduction, History, and Current Status. Vaccine Adjuv. 2017, 1494, 1–13. [Google Scholar]
- Harandi, A.M. Systems analysis of human vaccine adjuvants. Semin. Immunol. 2018, 39, 30–34. [Google Scholar] [CrossRef]
- Boyle, J.; Eastman, D.; Millar, C.; Camuglia, S.; Cox, J.; Pearse, M.; Good, J.; Drane, D. The utility of ISCOMATRIXTM adjuvant for dose reduction of antigen for vaccines requiring antibody responses. Vaccine 2007, 25, 2541–2544. [Google Scholar] [CrossRef]
- Brito, L.A.; Malyala, P.; O’Hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Vesikari, T.; Knuf, M.; Wutzler, P.; Karvonen, A.; Kieninger-Baum, D.; Schmitt, H.-J.; Baehner, F.; Borkowski, A.; Tsai, T.F.; Clemens, R. Oil-in-Water Emulsion Adjuvant with Influenza Vaccine in Young Children. N. Engl. J. Med. 2011, 365, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Podda, A. The adjuvanted influenza vaccines with novel adjuvants: experience with the MF59-adjuvanted vaccine. Vaccine 2001, 19, 2673–2680. [Google Scholar] [CrossRef]
- Khurana, S.; Chearwae, W.; Castellino, F.; Manischewitz, J.; King, L.R.; Honorkiewicz, A.; Rock, M.T.; Edwards, K.M.; Giudice, G.D.; Rappuoli, R.; et al. Vaccines with MF59 Adjuvant Expand the Antibody Repertoire to Target Protective Sites of Pandemic Avian H5N1 Influenza Virus. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Shi, Y. Alum: An old dog with new tricks. Emerg. Microbes Infect. 2016, 5, e25. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Didierlaurent, A.; Bourguignon, P.; Delhaye, S.; Baras, B.; Jacob, V.; Planty, C.; Elouahabi, A.; Harvengt, P.; Carlsen, H.; et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011, 29, 2461–2473. [Google Scholar] [CrossRef]
- Yam, K.K.; Gupta, J.; Winter, K.; Allen, E.; Brewer, A.; Beaulieu, É.; Mallett, C.P.; Burt, D.S.; Ward, B.J. AS03-Adjuvanted, Very-Low-Dose Influenza Vaccines Induce Distinctive Immune Responses Compared to Unadjuvanted High-Dose Vaccines in BALB/c Mice. Front. Immunol. 2015. [Google Scholar] [CrossRef]
- Galli, G.; Medini, D.; Borgogni, E.; Zedda, L.; Bardelli, M.; Malzone, C.; Nuti, S.; Tavarini, S.; Sammicheli, C.; Hilbert, A.K.; et al. Adjuvanted H5N1 vaccine induces early CD4+ T cell response that predicts long-term persistence of protective antibody levels. Proc. Natl. Acad. Sci. USA 2009, 106, 3877–3882. [Google Scholar] [CrossRef]
- Garçon, N.; Di Pasquale, A. From discovery to licensure, the Adjuvant System story. Hum. Vaccines Immunother. 2016, 13, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Garçon, N.; Vaughn, D.W.; Didierlaurent, A.M. Development and evaluation of AS03, an Adjuvant System containing α-tocopherol and squalene in an oil-in-water emulsion. Expert Rev. Vaccines 2012, 11, 349–366. [Google Scholar] [CrossRef]
- Bharucha, T.; Ming, D.; Breuer, J. A critical appraisal of ‘Shingrix’, a novel herpes zoster subunit vaccine (HZ/Su or GSK1437173A) for varicella zoster virus. Hum. Vaccines Immunother. 2017, 13, 1789–1797. [Google Scholar] [CrossRef]
- Sly, J.R.; Harris, A.L. Recombinant Zoster Vaccine (Shingrix) to Prevent Herpes Zoster. Nurs. Womens Health 2018, 22, 417–422. [Google Scholar] [CrossRef]
- Jalah, R.; Patel, V.; Kulkarni, V.; Rosati, M.; Alicea, C.; Ganneru, B.; von Gegerfelt, A.; Huang, W.; Guan, Y.; Broderick, K.E.; et al. IL-12 DNA as molecular vaccine adjuvant increases the cytotoxic T cell responses and breadth of humoral immune responses in SIV DNA vaccinated macaques. Hum. Vaccines Immunother. 2012, 8, 1620–1629. [Google Scholar] [CrossRef]
- Chong, S.-Y.; Egan, M.A.; Kutzler, M.A.; Megati, S.; Masood, A.; Roopchard, V.; Garcia-Hand, D.; Montefiori, D.C.; Quiroz, J.; Rosati, M.; et al. Comparative ability of plasmid IL-12 and IL-15 to enhance cellular and humoral immune responses elicited by a SIVgag plasmid DNA vaccine and alter disease progression following SHIV89.6P challenge in rhesus macaques. Vaccine 2007, 25, 4967–4982. [Google Scholar] [CrossRef]
- Khosroshahi, K.H.; Ghaffarifar, F.; Sharifi, Z.; D’Souza, S.; Dalimi, A.; Hassan, Z.M.; Khoshzaban, F. Comparing the effect of IL-12 genetic adjuvant and alum non-genetic adjuvant on the efficiency of the cocktail DNA vaccine containing plasmids encoding SAG-1 and ROP-2 of Toxoplasma gondii. Parasitol. Res. 2012, 111, 403–411. [Google Scholar] [CrossRef]
- Boyer, J.D.; Robinson, T.M.; Kutzler, M.A.; Parkinson, R.; Calarota, S.A.; Sidhu, M.K.; Muthumani, K.; Lewis, M.; Pavlakis, G.; Felber, B.; et al. SIV DNA vaccine co-administered with IL-12 expression plasmid enhances CD8 SIV cellular immune responses in cynomolgus macaques. J. Med. Primatol. 2005, 34, 262–270. [Google Scholar] [CrossRef]
- Sin, J.-I.; Kim, J.J.; Arnold, R.L.; Shroff, K.E.; McCallus, D.; Pachuk, C.; McElhiney, S.P.; Wolf, M.W.; Bruin, S.J.P.; Higgins, T.J.; et al. IL-12 Gene as a DNA Vaccine Adjuvant in a Herpes Mouse Model: IL-12 Enhances Th1-Type CD4+ T Cell-Mediated Protective Immunity Against Herpes Simplex Virus-2 Challenge. J. Immunol. 1999, 162, 2912–2921. [Google Scholar] [PubMed]
- Kim, J.J.; Ayyavoo, V.; Bagarazzi, M.L.; Chattergoon, M.A.; Dang, K.; Wang, B.; Boyer, J.D.; Weiner, D.B. In vivo engineering of a cellular immune response by coadministration of IL-12 expression vector with a DNA immunogen. J. Immunol. 1997, 158, 816–826. [Google Scholar] [PubMed]
- Kalams, S.A.; Parker, S.; Jin, X.; Elizaga, M.; Metch, B.; Wang, M.; Hural, J.; Lubeck, M.; Eldridge, J.; Cardinali, M.; et al. Safety and Immunogenicity of an HIV-1 Gag DNA Vaccine with or without IL-12 and/or IL-15 Plasmid Cytokine Adjuvant in Healthy, HIV-1 Uninfected Adults. PLoS ONE 2012, 7, e29231. [Google Scholar] [CrossRef]
- Kalams, S.A.; Parker, S.D.; Elizaga, M.; Metch, B.; Edupuganti, S.; Hural, J.; De Rosa, S.; Carter, D.K.; Rybczyk, K.; Frank, I.; et al. Safety and Comparative Immunogenicity of an HIV-1 DNA Vaccine in Combination with Plasmid Interleukin 12 and Impact of Intramuscular Electroporation for Delivery. J. Infect. Dis. 2013, 208, 818–829. [Google Scholar] [CrossRef]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Clavel, G.; Thiolat, A.; Boissier, M.-C. Interleukin newcomers creating new numbers in rheumatology: IL-34 to IL-38. Joint Bone Spine 2013, 80, 449–453. [Google Scholar] [CrossRef]
- Smith, D.E.; Renshaw, B.R.; Ketchem, R.R.; Kubin, M.; Garka, K.E.; Sims, J.E. Four New Members Expand the Interleukin-1 Superfamily. J. Biol. Chem. 2000, 275, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 Signal through IL-1Rrp2 and IL-1RAcP to Activate the Pathway Leading to NF-κB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef]
- Kumar, S.; McDonnell, P.C.; Lehr, R.; Tierney, L.; Tzimas, M.N.; Griswold, D.E.; Capper, E.A.; Tal-Singer, R.; Wells, G.I.; Doyle, M.L.; et al. Identification and Initial Characterization of Four Novel Members of the Interleukin-1 Family. J. Biol. Chem. 2000, 275, 10308–10314. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the interleukin-1 family of ligands and receptors. Semin. Immunol. 2013, 25, 389–393. [Google Scholar] [CrossRef]
- Dietrich, D.; Martin, P.; Flacher, V.; Sun, Y.; Jarrossay, D.; Brembilla, N.; Mueller, C.; Arnett, H.A.; Palmer, G.; Towne, J.; et al. Interleukin-36 potently stimulates human M2 macrophages, Langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 2016, 84, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Yazdi, A.S.; Ghoreschi, K. The Interleukin-1 Family. Regul. Cytokine Gene Expr. Immun. Dis. 2016, 941, 21–29. [Google Scholar]
- Vigne, S.; Palmer, G.; Martin, P.; Lamacchia, C.; Strebel, D.; Rodriguez, E.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ronchi, F.; et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 2012, 120, 3478–3487. [Google Scholar] [CrossRef]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Corbitt, N.; Pankhong, P.; Shin, T.; Khan, A.; Sardesai, N.Y.; Weiner, D.B. Immunogenicity of a novel engineered HIV-1 clade C synthetic consensus-based envelope DNA vaccine. Vaccine 2011, 29, 7173–7181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, V.L.; Patel, A.; Villarreal, D.O.; Hensley, S.E.; Ragwan, E.; Yan, J.; Sardesai, N.Y.; Rothwell, P.J.; Extance, J.P.; Caproni, L.J.; et al. Novel synthetic plasmid and DoggyboneTM DNA vaccines induce neutralizing antibodies and provide protection from lethal influenza challenge in mice. Hum. Vaccines Immunother. 2015, 11, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Griffin, B.D.; Agarwal, S.; Kudchodkar, S.B.; Reuschel, E.L.; Choi, H.; Kraynyak, K.A.; Duperret, E.K.; Keaton, A.A.; Chung, C.; et al. In vivo protection against ZIKV infection and pathogenesis through passive antibody transfer and active immunisation with a prMEnv DNA vaccine. NPJ Vaccines 2016, 1, 16021. [Google Scholar] [CrossRef]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) Ligands Require Processing for Full Agonist (IL-36α, IL-36β, and IL-36γ) or Antagonist (IL-36Ra) Activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef]
- Kumar, S.; Yan, J.; Muthumani, K.; Ramanathan, M.P.; Yoon, H.; Pavlakis, G.N.; Felber, B.K.; Sidhu, M.; Boyer, J.D.; Weiner, D.B. Immunogenicity Testing of a Novel Engineered HIV-1 Envelope Gp140 DNA Vaccine Construct. DNA Cell Biol. 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Choi, H.; Kudchodkar, S.B.; Reuschel, E.L.; Asija, K.; Borole, P.; Ho, M.; Wojtak, K.; Reed, C.; Ramos, S.; Bopp, N.E.; et al. Protective immunity by an engineered DNA vaccine for Mayaro virus. PLoS Negl. Trop. Dis. 2019, 13, e0007042. [Google Scholar] [CrossRef]
- Catalan-Dibene, J.; McIntyre, L.L.; Zlotnik, A. Interleukin 30 to Interleukin 40. J. Interferon Cytokine Res. 2018, 38, 423–439. [Google Scholar] [CrossRef]
- Debets, R.; Timans, J.C.; Homey, B.; Zurawski, S.; Sana, T.R.; Lo, S.; Wagner, J.; Edwards, G.; Clifford, T.; Menon, S.; et al. Two Novel IL-1 Family Members, IL-1δ and IL-1ε, Function as an Antagonist and Agonist of NF-κB Activation Through the Orphan IL-1 Receptor-Related Protein 2. J. Immunol. 2001, 167, 1440–1446. [Google Scholar] [CrossRef]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Clancy, D.M.; Henry, C.M.; Davidovich, P.B.; Sullivan, G.P.; Belotcerkovskaya, E.; Martin, S.J. Production of biologically active IL-36 family cytokines through insertion of N-terminal caspase cleavage motifs. FEBS Open Bio 2016, 6, 338–348. [Google Scholar] [CrossRef]
- André, S.; Seed, B.; Eberle, J.; Schraut, W.; Bültmann, A.; Haas, J. Increased Immune Response Elicited by DNA Vaccination with a Synthetic gp120 Sequence with Optimized Codon Usage. J. Virol. 1998, 72, 1497–1503. [Google Scholar] [Green Version]
- Deml, L.; Bojak, A.; Steck, S.; Graf, M.; Wild, J.; Schirmbeck, R.; Wolf, H.; Wagner, R. Multiple Effects of Codon Usage Optimization on Expression and Immunogenicity of DNA Candidate Vaccines Encoding the Human Immunodeficiency Virus Type 1 Gag Protein. J. Virol. 2001, 75, 10991–11001. [Google Scholar] [CrossRef] [Green Version]
- Wise, M.C.; Hutnick, N.A.; Pollara, J.; Myles, D.J.F.; Williams, C.; Yan, J.; LaBranche, C.C.; Khan, A.S.; Sardesai, N.Y.; Montefiori, D.; et al. An Enhanced Synthetic Multiclade DNA Prime Induces Improved Cross-Clade-Reactive Functional Antibodies when Combined with an Adjuvanted Protein Boost in Nonhuman Primates. J. Virol. 2015, 89, 9154. [Google Scholar] [CrossRef]
- Ding, L.; Wang, X.; Hong, X.; Lu, L.; Liu, D. IL-36 cytokines in autoimmunity and inflammatory disease. Oncotarget 2017, 9, 2895–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 promotes myeloid cell infiltration, activation and inflammatory activity in skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef]
- Traks, T.; Keermann, M.; Prans, E.; Karelson, M.; Loite, U.; Kõks, G.; Silm, H.; Kõks, S.; Kingo, K. Polymorphisms in IL36G gene are associated with plaque psoriasis. BMC Med. Genet. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Black, G.C.M.; Clayton, T.H.; Judge, M.; Griffiths, C.E.M.; Warren, R.B. A novel mutation in IL36RN underpins childhood pustular dermatosis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 302–305. [Google Scholar] [CrossRef]
- Mahil, S.K.; Catapano, M.; Meglio, P.D.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med. 2017, 9, eaan2514. [Google Scholar] [CrossRef] [PubMed]
- Ainscough, J.S.; Macleod, T.; McGonagle, D.; Brakefield, R.; Baron, J.M.; Alase, A.; Wittmann, M.; Stacey, M. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc. Natl. Acad. Sci. USA 2017, 114, E2748–E2757. [Google Scholar] [CrossRef]
- Meier-Schiesser, B.; Feldmeyer, L.; Jankovic, D.; Mellett, M.; Satoh, T.K.; Yerly, D.; Navarini, A.; Abe, R.; Yawalkar, N.; Chung, W.-H.; et al. Culprit Drugs Induce Specific IL-36 Overexpression in Acute Generalized Exanthematous Pustulosis. J. Investig. Dermatol. 2019, 139, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.-A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.-C.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Di Caprio, R.; Balato, A.; Caiazzo, G.; Lembo, S.; Raimondo, A.; Fabbrocini, G.; Monfrecola, G. IL-36 cytokines are increased in acne and hidradenitis suppurativa. Arch. Dermatol. Res. 2017, 309, 673–678. [Google Scholar] [CrossRef]
- Towne, J.; Sims, J. IL-36 in psoriasis. Curr. Opin. Pharmacol. 2012, 12, 486–490. [Google Scholar] [CrossRef]
- Hashiguchi, Y.; Yabe, R.; Chung, S.-H.; Murayama, M.A.; Yoshida, K.; Matsuo, K.; Kubo, S.; Saijo, S.; Nakamura, Y.; Matsue, H.; et al. IL-36α from Skin-Resident Cells Plays an Important Role in the Pathogenesis of Imiquimod-Induced Psoriasiform Dermatitis by Forming a Local Autoamplification Loop. J. Immunol. 2018, 201, 167–182. [Google Scholar] [CrossRef]
- Wang, W.; Yu, X.; Wu, C.; Jin, H. IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis. Int. J. Med. Sci. 2017, 14, 1002–1007. [Google Scholar] [CrossRef]
- Kanazawa, N.; Nakamura, T.; Mikita, N.; Furukawa, F. Novel IL36RN mutation in a Japanese case of early onset generalized pustular psoriasis. J. Dermatol. 2013, 40, 749–751. [Google Scholar] [CrossRef]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schäfer, M.; Coyle, A.J.; Renauld, J.-C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36–mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Arakawa, A.; Vollmer, S.; Besgen, P.; Galinski, A.; Summer, B.; Kawakami, Y.; Wollenberg, A.; Dornmair, K.; Spannagl, M.; Ruzicka, T.; et al. Unopposed IL-36 Activity Promotes Clonal CD4+ T-Cell Responses with IL-17A Production in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2018, 138, 1338–1347. [Google Scholar] [CrossRef]
- Verma, A.H.; Zafar, H.; Ponde, N.O.; Hepworth, O.W.; Sihra, D.; Aggor, F.E.Y.; Ainscough, J.S.; Ho, J.; Richardson, J.P.; Coleman, B.M.; et al. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J. Immunol. 2018, 201, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Winkle, S.M.; Throop, A.L.; Herbst-Kralovetz, M.M. IL-36γ Augments Host Defense and Immune Responses in Human Female Reproductive Tract Epithelial Cells. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Gardner, J.K.; Herbst-Kralovetz, M.M. IL-36γ induces a transient HSV-2 resistant environment that protects against genital disease and pathogenesis. Cytokine 2018, 111, 63–71. [Google Scholar] [CrossRef]
- Kovach, M.A.; Singer, B.; Martinez-Colon, G.; Newstead, M.W.; Zeng, X.; Mancuso, P.; Moore, T.A.; Kunkel, S.L.; Peters-Golden, M.; Moore, B.B.; et al. IL-36γ is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal. Immunol. 2017, 10, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Milora, K.A.; Uppalapati, S.R.; Sanmiguel, J.C.; Zou, W.; Jensen, L.E. Interleukin-36β provides protection against HSV-1 infection, but does not modulate initiation of adaptive immune responses. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Aoyagi, T.; Newstead, M.W.; Zeng, X.; Nanjo, Y.; Peters-Golden, M.; Kaku, M.; Standiford, T.J. Interleukin-36γ and IL-36 receptor signaling mediate impaired host immunity and lung injury in cytotoxic Pseudomonas aeruginosa pulmonary infection: Role of prostaglandin E2. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Feng, C.; Weinstein, A.; Xia, R.; Wen, W.; Lv, Q.; Zuo, S.; Tang, P.; Yang, X.; et al. IL-36γ transforms the tumor microenvironment and promotes type 1 lymphocyte-mediated antitumor immune responses. Cancer Cell 2015, 28, 296. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louis, L.; Wise, M.C.; Choi, H.; Villarreal, D.O.; Muthumani, K.; Weiner, D.B. Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model. Vaccines 2019, 7, 42. https://doi.org/10.3390/vaccines7020042
Louis L, Wise MC, Choi H, Villarreal DO, Muthumani K, Weiner DB. Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model. Vaccines. 2019; 7(2):42. https://doi.org/10.3390/vaccines7020042
Chicago/Turabian StyleLouis, Lumena, Megan C. Wise, Hyeree Choi, Daniel O. Villarreal, Kar Muthumani, and David B. Weiner. 2019. "Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model" Vaccines 7, no. 2: 42. https://doi.org/10.3390/vaccines7020042
APA StyleLouis, L., Wise, M. C., Choi, H., Villarreal, D. O., Muthumani, K., & Weiner, D. B. (2019). Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model. Vaccines, 7(2), 42. https://doi.org/10.3390/vaccines7020042