Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa



Abstract
:1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis of African Strains of the Newcastle Disease Virus
2.1.1. RNA Isolation, cDNA Synthesis, and Nucleotide Sequencing
2.1.2. Accession Numbers and Nucleotide Sequences for Gene Cloning
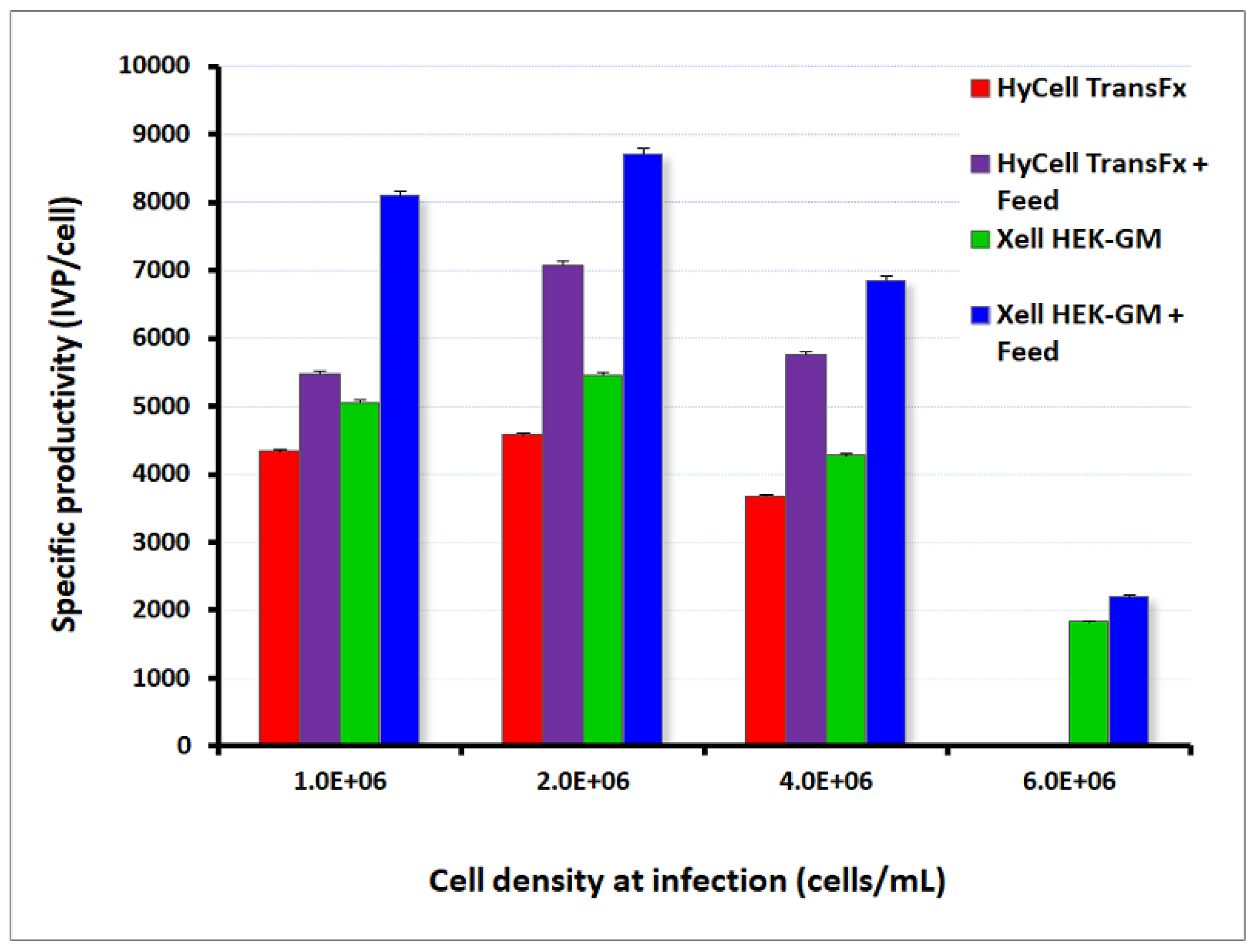
2.2. Cells Lines and Culture Media
2.3. Adenovirus Vectors Design, Rescue, and Seed-Stock Generation
2.3.1. Adenoviral Vectors Design
2.3.2. Adenovirus Rescue
2.4. Analytical Assays and Characterization of the Recombinant Adenoviruses
2.4.1. Cell Count and Viability
2.4.2. Total Particle Quantitation of Adenovectors
2.4.3. Infectious Particle Titration
2.4.4. Enzyme-Linked Immunosorbent Assays (ELISA) and Hemagglutination Inhibition Assay (HIA)
2.4.5. PCR Analyses and Sequencing
2.5. Cell Culture and Upstream Process Development for Adenoviral Vector Production
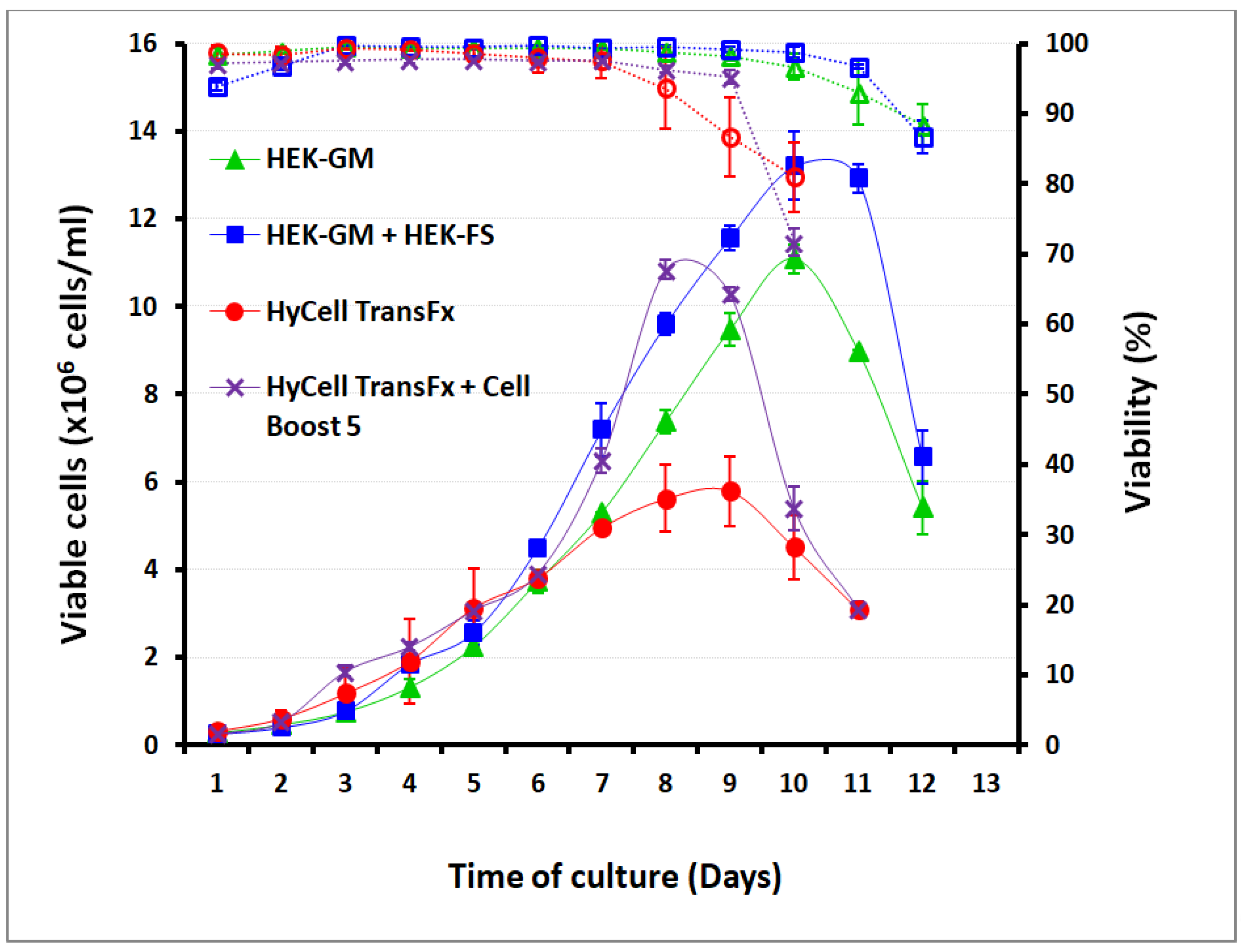
2.5.1. Cell Growth Kinetics and Media Performances
2.5.2. Virus Production at Small Scale
2.6. Bioreactors, Operating Conditions and Online Data Processing
2.7. Purification of the Recombinant Adenoviruses
2.7.1. Cells Harvest, Lysis, and Free Nucleic Acid Digestion
2.7.2. Purification by CsCl Ultracentrifugation
2.8. Animals, Immunization Experiments, and Viral Challenge
2.8.1. Ethics Statement
2.8.2. Vaccination and Challenge NDV Strains
2.8.3. Mice Experiment
2.8.4. Target animal experiment
2.9. Statistical Analyses
3. Results
3.1. Antigenically Matched F and HN Antigens for Vaccine Design
3.2. Construction of Adenoviral Vectors Carrying the Foreign F and HN Gene Sequences from NDV
3.3. Adenovirus Production and Operating Culture Conditions in Shake Flasks
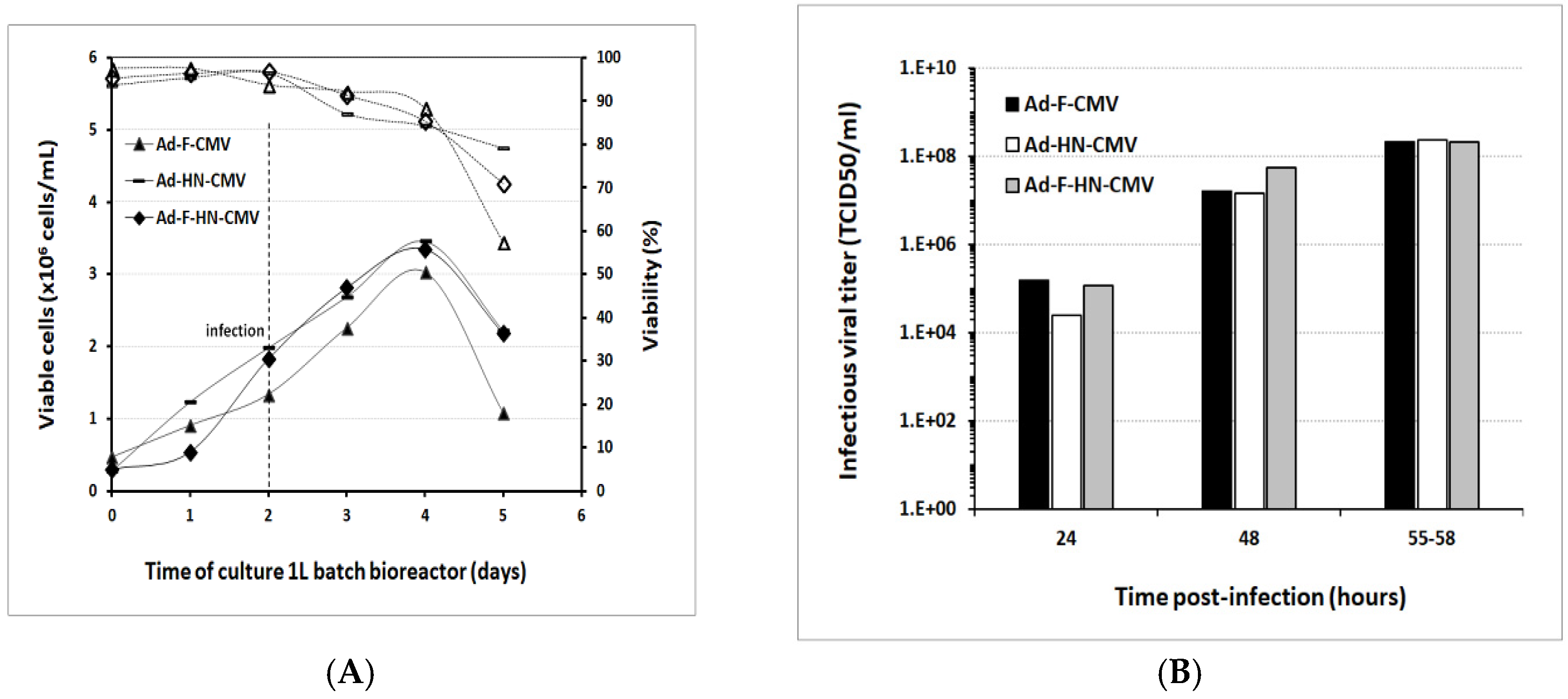
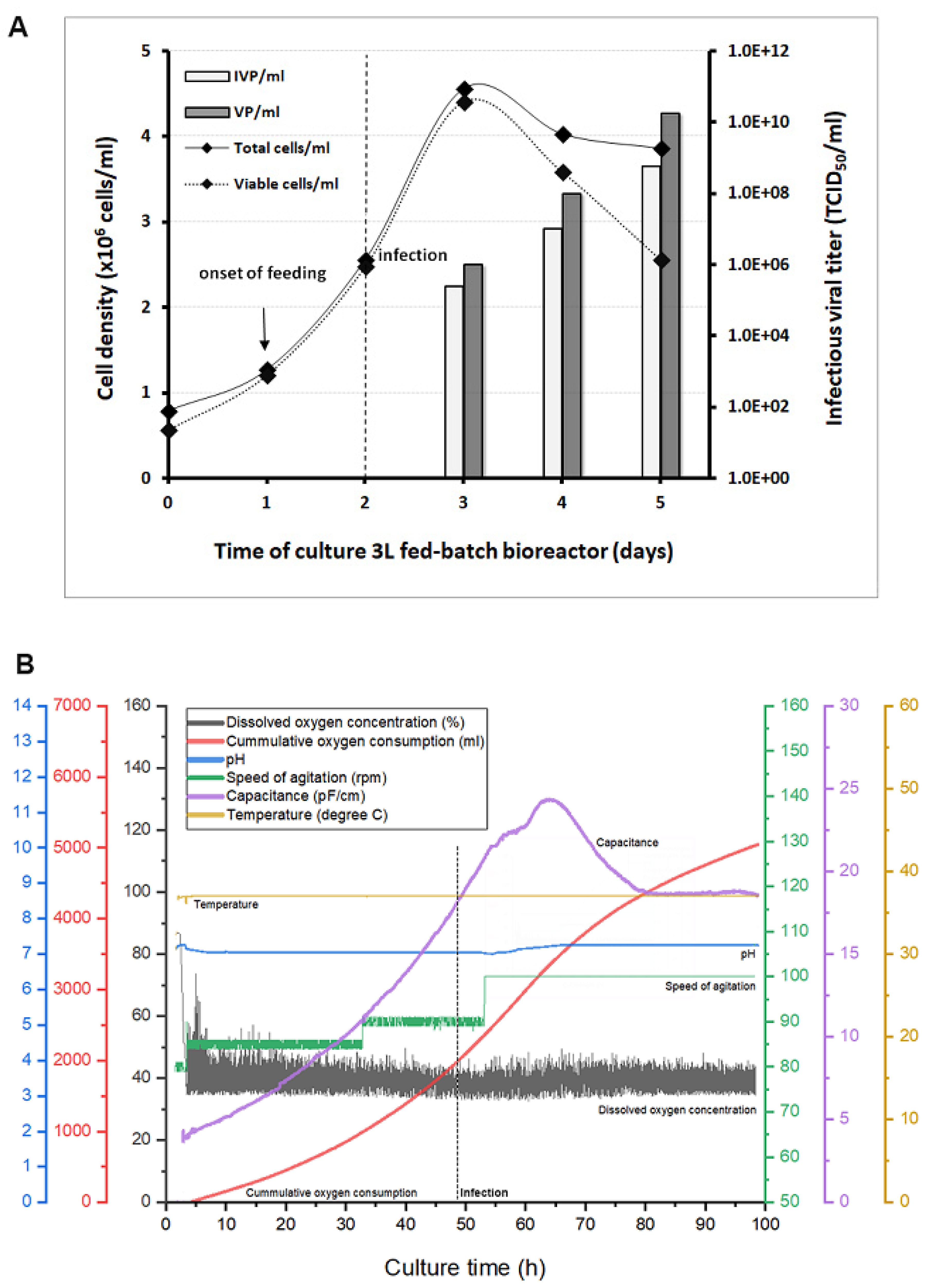
3.4. Scale-Up for Adenovirus Production In 1 and 3 L Bioreactors
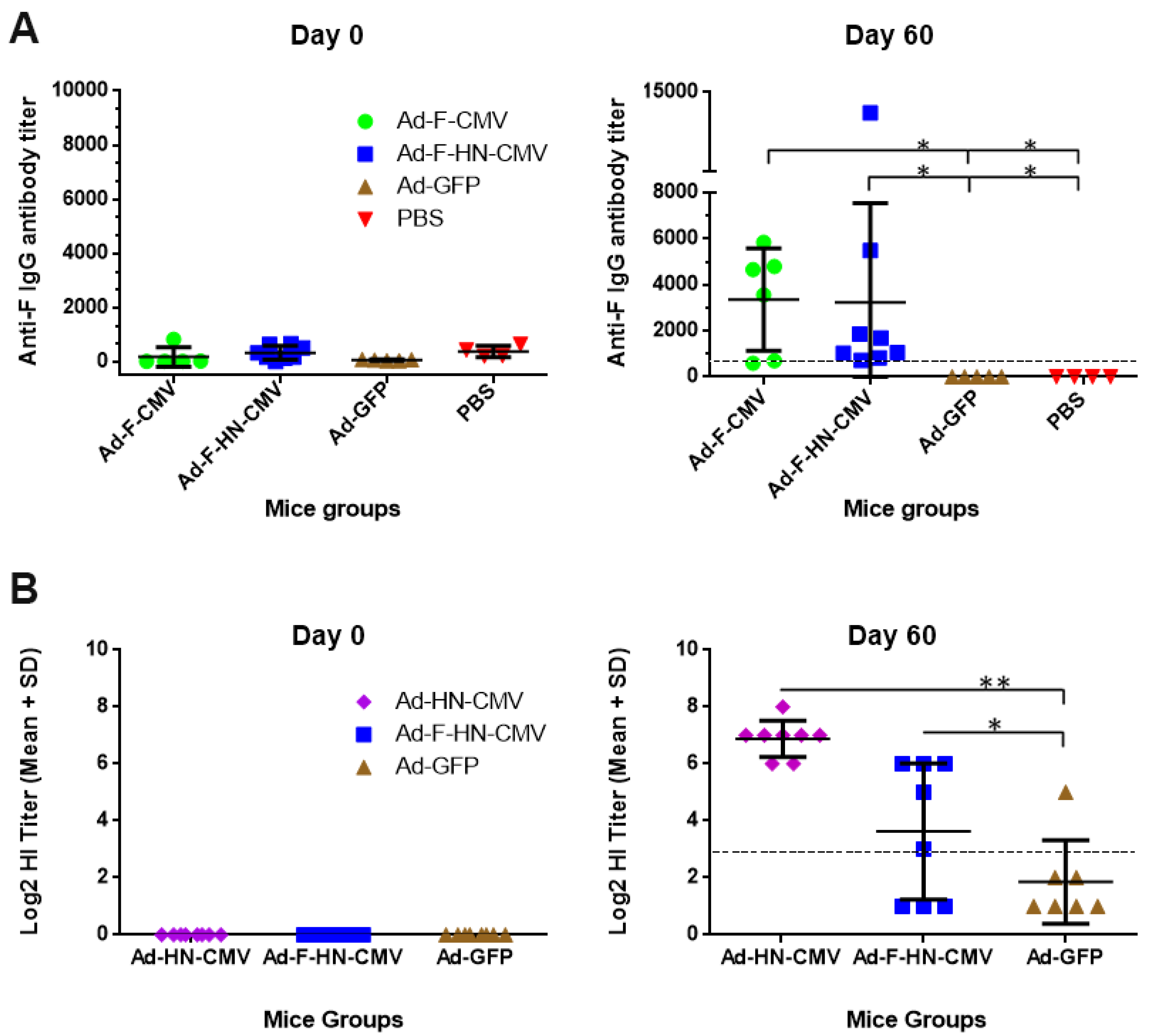
3.5. Immunogenicity and Protective Efficacy Assessments of the NDV Adenovirus Vaccine Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Grace, D.; Roesel, K.; Lore, T. Poverty and Gender Aspects of Food Safety and Informal Markets in Sub-Saharan Africa; ILRI (aka ILCA and ILRAD): Nairobi, Kenya, 2014. [Google Scholar]
- Dimitrov, K.M.; Afonso, C.L.; Yu, Q.; Miller, P.J. Newcastle disease vaccines—A solved problem or a continuous challenge? Vet. Microbiol. 2017, 206, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, C.; Adeyanju, A.T.; Owoade, A.A.; Couacy-Hymann, E.; Alkali, B.R.; Ottosson, U.; Muller, C.P. Genetic Diversity of Newcastle Disease Virus in Wild Birds and Pigeons in West Africa. Appl. Environ. Microbiol. 2013, 79, 7867–7874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, P.J.; Afonso, C.L.; El Attrache, J.; Dorsey, K.M.; Courtney, S.C.; Guo, Z.; Kapczynski, D.R. Effects of Newcastle Disease Virus Vaccine Antibodies on the Shedding and Transmission of Challenge Viruses. Dev. Comp. Immunol. 2013, 41, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degefa, T.; Dadi, L.; Yami, A.; GMariam, K.; Nassir, M. Technical and economic evaluation of different methods of Newcastle disease vaccine administration. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2004, 51, 365–369. [Google Scholar] [CrossRef]
- Shittu, I.; Zhu, Z.; Lu, Y.; Hutcheson, J.M.; Stice, S.L.; West, F.D.; Donadeu, M.; Dungu, B.; Fadly, A.M.; Zavala, G.; et al. Development, Characterization and Optimization of a New Suspension Chicken-Induced Pluripotent Cell Line for the Production of Newcastle Disease Vaccine. Biologicals 2016, 44, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.F. Vaccine preparedness—Are we ready for the next influenza pandemic? N. Engl. J. Med. 2008, 358, 2540–2543. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.T. Poultry production and performance in the Federal Democratic Republic of Ethiopia. World Poult. Sci. J. 2010, 66, 441–454. [Google Scholar] [CrossRef]
- OIE. Newcastle disease. In OIE Manual for Diagnostic Tests and Vaccines for Terrestrial Animals, 5th ed.; OIE: Paris, France, 2004; Volume 1, pp. 270–283. [Google Scholar]
- Allan, W.H.; Lancaster, J.E.; Toth, B. Newcastle disease vaccines—Their production and use. FAO Anim. Prod. Ser. Rome 1978, 10, 1–163. [Google Scholar]
- Alexander, D.J. Newcastle disease virus—An avian Paramyxovirus. In Newcastle Disease; Alexander, D.J., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1988; pp. 11–22. [Google Scholar]
- Alexander, D.J.; Senne, D.A. Newcastle Disease, other avian paramyxoviruses, and pneumovirus infections. In Diseases of Poultry, 12th ed.; Saif, Y.M., Fadly, A.M., Glisson, J.R., McDougald, L.R., Nolan, L.K., Swayne, D.E., Eds.; Iowa State University Press: Ames, IA, USA, 2008; pp. 75–116. [Google Scholar]
- Kim, S.H.; Wanasen, N.; Paldurai, A.; Xiao, S.; Collins, P.L.; Samal, S.K. Newcastle disease virus fusion protein is the major contributor to protective immunity of genotype-matched vaccine. PLoS ONE 2013, 8, e74022. [Google Scholar] [CrossRef]
- Miller, P.J.; Koch, G. Newcastle disease. In Diseases of Poultry; Swayne, D.E., Glisson, J.R., McDougald, L.R., Nolan, L.K., Suarez, D.L., Nair, V., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 89–138. [Google Scholar]
- Steward, M.; Vipond, I.B.; Millar, N.S.; Emmerson, P.T. RNA editing in Newcastle disease virus. J. Gen. Virol. 1993, 74, 2539–2547. [Google Scholar] [CrossRef]
- Dortmans, J.C.; Koch, G.; Rottier, P.J.; Peeters, B.P. Virulence of newcastle disease virus: What is known so far? Vet. Res. 2011, 42, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diel, D.G.; da Silva, L.H.; Liu, H.; Wang, Z.; Miller, P.J.; Afonso, C.L. Genetic diversity of avian paramyxovirus type 1: Proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infect. Genet. Evol. 2012, 12, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Courtney, S.C.; Susta, L.; Gomez, D.; Hines, N.; Pearson, J.E.; Brown, C.C.; Miller, P.J.; Afonso, C.L. Highly divergent virulent isolates of Newcastle disease virus from the Dominican Republic are members of a new genotype that may have evolved unnoticed for over two decades. J. Clin. Microbiol. 2012, 51, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, K.; Ying, G.; Yan, Z.; Shanshan, Y.; Lei, Z.; Hongjun, L.; Maosheng, S. Progress on adenovirus-vectored universal influenza vaccines. Hum. Vaccines Immunother. 2015, 11, 1209–1222. [Google Scholar] [CrossRef] [Green Version]
- Kallel, H.; Kamen, A.A. Large-scale adenovirus and poxvirus-vectored vaccine manufacturing to enable clinical trials. Biotechnol. J. 2015, 10, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Preparation of Recombinant Human Adenoviruses Labeled With miniSOG. Available online: https://pubmed.ncbi.nlm.nih.gov/27295881/ (accessed on 26 June 2020).
- Fernandez-Sainz, I.; Medina, G.N.; Ramirez-Medina, E.; Koster, M.J.; Grubman, M.J.; de Los Santos, T. Adenovirus-vectored Foot-And-Mouth Disease Vaccine Confers Early and Full Protection Against FMDV O1 Manisa in Swine. Virology 2017, 502, 123–132. [Google Scholar] [CrossRef]
- Pedersen, L.E.; Patch, J.R.; Kenney, M.; Glabman, R.A.; Nielsen, M.; Jungersen, G.; Buus, S.; Golde, W.T. Expanding specificity of class I restricted CD8+ T cells for viral epitopes following multiple inoculations of swine with a human adenovirus vectored foot-and-mouth disease virus (FMDV) vaccine. Vet. Immunol. Immunopathol. 2016, 181, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoke, C.H.; Snyder, C.E. History of the restoration of adenovirus type 4 and type 7 vaccine, live oral (Adenovirus Vaccine) in the context of the Department of Defense acquisition system. Vaccine 2013, 31, 1623–1632. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Cote, J.; Garnier, A.; Massie, B.; Kamen, A. Serum-free production of recombinant proteins and adenoviral vectors by 293SF-3F6 cells. Biotechnol. Bioeng. 1998, 59, 567–575. [Google Scholar] [CrossRef]
- Stillman, B.W.; Gluzman, Y. Replication and supercoiling of simian virus 40 DNA in cell extracts from human cells. Mol. Cell. Biol. 1985, 5, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- AdEasy Adenoviral Vector System Instructions Manual. Available online: https://www.agilent.com/cs/library/usermanuals/public/240009.pdf (accessed on 24 June 2020).
- Kamen, A.; Henry, O. Development and optimization of an adenovirus production process. J. Gene Med. 2004, 6, S184–S192. [Google Scholar] [CrossRef] [PubMed]
- Kangro, H.; Mahy, B. Virology Methods Manual, 1st ed.; Academic Press: Cambridge, MA, USA, 1996. [Google Scholar]
- Abdi, R.D.; Amsalu, K.; Merera, O.; Asfaw, Y.; Gelaye, E.; Yami, Y.; Sori, T. Serological response and protection level evaluation in chickens exposed to grains coated with I2 Newcastle disease virus for effective oral vaccination of village chickens. BMC Vet. Res. 2016, 12, 150. [Google Scholar]
- Alexander, D.J.; Aldous, E.W.; Fuller, C.M. The long view: A selective review of 40 years of Newcastle disease research. Avian Pathol. 2012, 41, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kaleta, E.F.; Baldauf, C. Newcastle disease in free-living and pet birds. In Newcastle Disease; Alexander, D.J., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, MA, USA, 1988; pp. 197–246. [Google Scholar]
- World Bank; TAFS Forum. World Livestock Disease Atlas. A Quantitative Analysis of Global Animal Health Data (2006–2009), Forum 2011; World Bank: Washington, DC, USA, 2011. [Google Scholar]
- Duguma, R. Understanding the role of indigenous chicken during the long walk to food security in Ethiopia. LRRD 2009, 21, 116. [Google Scholar]
- Palya, V. Manual for the production of Marek’s disease, Gumboro disease and inactivated Newcastle disease vaccines. Technical Centre for Agricultural and Rural Cooperation. FAO Anim. Prod. Health Pap. 1991, 89, 1–25. [Google Scholar]
- Marangon, S.; Busani, L. The use of vaccination in poultry production. Rev. Sci. Tech. Off. Int. Epizoot. 2006, 26, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.H.; Kwon, H.J.; Kim, T.E.; Kim, J.H.; Yoo, H.S.; Park, M.H.; Park, Y.H.; Kim, S.J. Characterization of a recombinant Newcastle disease vaccine strain. Clin. Vaccine Immunol. 2008, 15, 1572–1579. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.J.; King, D.J.; Afonso, C.L.; Suarez, D.L. Antigenic differences among Newcastle disease virus strains of different genotypes used in vaccine formulation affect viral shedding after a virulent challenge. Vaccine 2007, 25, 7238–7246. [Google Scholar] [CrossRef]
- Xiao, S.; Nayak, B.; Samuel, A.; Paldurai, A.; Kanabagattebasavarajappa, M.; Prajitno, T.Y.; Bharoto, E.E.; Collins, P.L.; Samal, S.K. Generation by reverse genetics of an effective, stable, live-attenuated Newcastle disease virus vaccine based on a currently circulating, highly virulent Indonesian strain. PLoS ONE 2012, 7, e52751. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.J.; Estevez, C.; Yu, Q.; Suarez, D.L.; King, D.J. Comparison of viral shedding following vaccination with inactivated and live Newcastle disease vaccines formulated with wild-type and recombinant viruses. Avian Dis. 2009, 53, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.W.; Yang, H.M.; Jin, J.H.; Zhao, J.; Xue, J.; Zhang, G.Z. Recombinant Newcastle disease virus (NDV) La Sota expressing the hemagglutinin–neuraminidase protein of genotype VII NDV shows improved protection efficacy against NDV challenge. Avian Pathol. 2019, 48, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Afrough, S.; Rhodes, S.; Evans, T.; White, R.; Benest, J. Immunologic Dose-Response to Adenovirus-Vectored Vaccines in Animals and Humans: A Systematic Review of Dose-Response Studies of Replication Incompetent Adenoviral Vaccine Vectors When Given via an Intramuscular or Subcutaneous Route. Vaccines 2020, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fougeroux, C.; Holst, P.J. Future Prospects for the Development of Cost-Effective Adenovirus Vaccines. Int. J. Mol. Sci. 2017, 18, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.F.; Lanthier, S.; Jacob, D.; Montes, J.; Beath, A.; Beresford, A.; Kamen, A. Process optimization and scale-up for production of rabies vaccine live adenovirus vector (AdRG1.3). Vaccine 2012, 30, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.F.; Jacob, D.; Zhuc, T.; Bernier, A.; Shao, Z.; Yub, X.; Patela, M.; Lanthiera, S.; Kamen, A. Optimization and scale-up of cell culture and purification processes for production of an adenovirus-vectored tuberculosis vaccine candidate. Vaccine 2016, 34, 3381–3387. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.D.; Iqbal, M.; Nair, V. Recent advances in viral vectors in veterinary vaccinology. Curr. Opin. Virol. 2018, 29, 1–7. [Google Scholar] [CrossRef]
- Gong, X.; Li, D.; Li, X.; Fang, Q.; Han, X.; Wu, Y.; Yang, S.; Shen, B.Q. Fed-batch culture optimization of a growth-associated hybridoma cell line in chemically defined protein-free media. Cytotechnology 2006, 52, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, S. A new approach for finding smooth optimal feeding profiles in fed-batch fermentations. Biochem. Eng. J. 2016, 105, 177–188. [Google Scholar] [CrossRef]
- Chen, S.; Guo, D.; Guo, B.; Liu, J.; Shen, Y.; Xu, X.; Huang, W.; Guo, S. Investigation on Formulation and Preparation of Adenovirus Encoding Human Endostatin Lyophilized Powders. Int. J. Pharm. 2012, 427, 145–152. [Google Scholar] [CrossRef]
- Gilbert, A.; Johnson, S.; Walker, N.; Wickham, C.; Beath, A.; VerCauteren, K. Efficacy of Ontario Rabies Vaccine Baits (ONRAB) against rabies infection in raccoons. Vaccine 2018, 36, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Zhu, T.; Wu, S.; Feng, L.; Cheng, P.; Liu, J.; Wang, J. Establishing China’s national standard for virus titer of the recombinant adenovirus type-5 vector-based Ebola vaccine (Ad5-EBOV). Hum. Gene Ther. Clin. Dev. 2018, 29, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.B.; Alves, P.M.; Aunins, J.G.; Carrondo, M.J.T. Use of adenoviral vectors as veterinary vaccines. Gene Ther. 2005, 12, S73–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Garib, S.O.; Gielkens, A.L.J.; Gruys, D.E.; Hartog, L.; Koch, G. Immunoglobulin Class Distribution of Systemic and Mucosal Antibody Responses to Newcastle Disease in Chickens. Avian Dis. 2003, 47, 32–40. [Google Scholar] [CrossRef]
- Kapczynski, D.R.; Afonso, C.L.; Miller, P.J. Immune responses of poultry to Newcastle disease virus. Dev. Comp. Immunol. 2013, 41, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture Mode in Bioreactor | Batch 1 L | Batch 1 L | Batch 1 L |
|---|---|---|---|
| Adenovirus produced | Ad-F-CMV | Ad-HN-CMV | Ad-F-HN-CMV |
| Peak of cell density in the production phase | 3.42 × 106 cells/mL | 4.01 × 106 cells/mL | 3.93 × 106 cells/mL |
| Total cell density at harvest | 1.88 × 106 cells/mL | 2.76 × 106 cells/mL | 3.08 × 106 cells/mL |
| Viability at harvest | 57.4% | 79.1% | 71.0% |
| Total viral particle concentration in cell culture lysate supernatants | 4.80 × 1010 VP/mL | 3.45 × 1010 VP/mL | 2.64 × 1010 VP/mL |
| Infectious viral particle concentration in cell culture lysate supernatants | 2.17 × 108 IVP/mL | 1.29 × 108 IVP/mL | 1.80 × 108 IVP/mL |
| IVP/VP ratio in cell culture lysate supernatants | ~0.4% | ~0.4% | ~0.7% |
| Total viral particle concentration in CsCl purified samples | 1.08 × 1011 VP/mL | 1.02 × 1011 VP/mL | 1.0 × 1011 VP/mL |
| Infectious viral particle concentration in CsCl purified samples | 3.21 × 109 IVP/mL | 4.06 × 109 IVP/mL | 1.86 × 109 IVP/mL |
| IVP/VP in CsCl purified, concentrated vaccine stocks | ~3% | ~4% | ~2% |
| Culture Mode in Bioreactor | Fed-Batch 1 L | Fed-Batch 1 L | Fed-Batch 1 L | Fed-Batch 3 L | |
|---|---|---|---|---|---|
| Adenovirus produced | Ad-F-CMV (1) | Ad-F-CMV (2) | Ad-F-βactin | Ad-F-HN-CMV | Ad-F-βactin |
| Peak of cell density in the production phase | 4.28 × 106 cells/mL | 4.35 × 106 cells/mL | 3.87 × 106 cells/mL | 4.56 × 106 cells/mL | 3.81 × 106 cells/mL |
| Total cell density at harvest | 4.13 × 106 cells/mL | 4.18 × 106 cells/mL | 3.08 × 106 cells/mL | 3.86 × 106 cells/mL | 1.86 × 106 cells/mL |
| Viability at harvest | 78.4% | 80.1% | 60.7% | 66.5% | 76.2% |
| Total viral particle concentration in cell culture lysate supernatants | 1.08 × 1010 VP/mL | 2.24 × 1010 VP/mL | 1.19 × 1010 VP/mL | 1.78 × 1010 VP/mL | 1.58 × 1010 VP/mL |
| Infectious viral particle concentration in cell culture lysate supernatants | 5.01 × 108 IVP/mL | 5.64 × 108 IVP/mL | 3.2 × 108 IVP/mL | 5.80 × 108 IVP/mL | 5.03 × 108 IVP/mL |
| IVP/VP ratio in cell culture lysate supernatants | ~5% | ~2.5% | ~2.7% | ~3.3% | ~3.2% |
| Total viral particle concentration in CsCl purified samples | 7.01 × 1010 VP/mL | 5.23 × 1010 VP/mL | 1.36 × 1011 VP/mL | 5.35 × 1011 VP/mL | 1.11 × 1011 VP/mL |
| Infectious viral particle concentration in CsCl purified samples | 1.03 × 1010 IVP/mL | 1.06 × 1010 IVP/mL | 3.2 × 1010 IVP/mL | 6.30 × 1010 IVP/mL | 2.46 × 1010 IVP/mL |
| IVP/VP ratio in CsCl purified, concentrated vaccine stocks | ~15% | ~20% | ~22% | ~12% | ~22% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farnós, O.; Gelaye, E.; Trabelsi, K.; Bernier, A.; Subramani, K.; Kallel, H.; Yami, M.; Kamen, A.A. Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa. Vaccines 2020, 8, 338. https://doi.org/10.3390/vaccines8020338
Farnós O, Gelaye E, Trabelsi K, Bernier A, Subramani K, Kallel H, Yami M, Kamen AA. Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa. Vaccines. 2020; 8(2):338. https://doi.org/10.3390/vaccines8020338
Chicago/Turabian StyleFarnós, Omar, Esayas Gelaye, Khaled Trabelsi, Alice Bernier, Kumar Subramani, Héla Kallel, Martha Yami, and Amine A. Kamen. 2020. "Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa" Vaccines 8, no. 2: 338. https://doi.org/10.3390/vaccines8020338
APA StyleFarnós, O., Gelaye, E., Trabelsi, K., Bernier, A., Subramani, K., Kallel, H., Yami, M., & Kamen, A. A. (2020). Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa. Vaccines, 8(2), 338. https://doi.org/10.3390/vaccines8020338