SARS-CoV-2 Consensus-Sequence and Matching Overlapping Peptides Design for COVID19 Immune Studies and Vaccine Development

 , , ,
, , ,
Abstract
1. Introduction
2. Methods
2.1. Consensus Sequence ORF Generation and Entropy Calculation
2.2. Overlapping Peptide Set Design and Variability Plots
2.3. Detection of Conserved Peptides Among Coronavirus
2.4. Identification of Previously Described Epitopes in CoV-2 Conserved Regions
3. Results
3.1. Open Reading Frames and Sequence Isolates for CoV-2-Cons Sequence Creation
3.2. Overlapping Peptides (OLP) Sets Design
3.3. CoV-2-Cons Variability Analysis by Entropy Scores across the Full Genome
3.4. Variant OLP Sequences to Cover CoV-2 Sequence Diversity
3.5. Conserved Protein Sequences Matching Other Coronavirus Family Member and Identification of Pan-Coronavirus Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Channappanavar, R.; Zhao, J.; Perlman, S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014, 59, 118–128. [Google Scholar] [CrossRef]
- Zhao, J.; Alshukairi, A.N.; Baharoon, S.A.; Ahmed, W.A.; Bokhari, A.A.; Nehdi, A.M.; Layqah, L.A.; Alghamdi, M.G.; Al Gethamy, M.M.; Dada, A.M.; et al. Recovery from the Middle East respiratory syndrome is associated with antibody and T cell responses. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhao, M.; Liu, K.; Xu, K.; Wong, G.; Tan, W.; Gao, G.F. T-cell immunity of SARS-CoV: Implications for vaccine development against MERS-CoV. Antiviral Res. 2017, 137, 82–92. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 1–10. [Google Scholar] [CrossRef]
- Wu, F.; Wang, A.; Liu, M.; Wang, Q.; Chen, J.; Xia, S.; Ling, Y.; Zhang, Y.; Xun, J.; Lu, L.; et al. Neutralizing Antibody Responses to SARS-CoV-2 in a COVID-19 Recovered Patient Cohort and Their Implications. medRxiv 2020. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Straling, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Robbiani, D.; Gaebler, C.; Muecksch, F.; Lorenzi, J.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.; Gazumyan, A.; Finkin, S.; et al. Convergent Antibody Responses to SARS-CoV-2 Infection in Convalescent Individuals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4 + and CD8 + T cells induced by SARS-CoV-2 in UK convalescent COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Rydyznski Moderbacher, C.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Journal Pre-Proof Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020, 181. [Google Scholar] [CrossRef] [PubMed]
- Gallais, F.; Velay, A.; Wendling, M.-J.; Nazon, C.; Partisani, M.; Sibilia, J.; Candon, S.; Fafi-Kremer, S. Intrafamilial Exposure to SARS-CoV-2 Induces Cellular Immune Response without Seroconversion. medRxiv 2020. [Google Scholar] [CrossRef]
- Seow, J.; Graham, C.; Merrick, B.; Acors, S.; Steel, K.J.A.; Hemmings, O.; O’Bryne, A.; Kouphou, N.; Pickering, S.; Galao, R.; et al. Longitudinal evaluation and decline of antibody responses in SARS-CoV-2 infection. medRxiv 2020. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020, 92, 495–500. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B.C.; Lee, S.S.; Chakraborty, C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med. Virol. 2020, 92, 618–631. [Google Scholar] [CrossRef]
- Gao, A.; Chen, Z.; Segal, F.P.; Carrington, M.M.; Streeck, H.; Chakraborty, A.K.; Juelg, B.; Julg, B. Predicting the Immunogenicity of T cell epitopes: From HIV to SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680.e2. [Google Scholar] [CrossRef]
- Lucchese, G. Epitopes for a 2019-nCoV vaccine. Cell. Mol. Immunol. 2020, 17, 539–540. [Google Scholar] [CrossRef]
- Silva-Arrieta, S.; Goulder, P.J.R.; Brander, C. In silico veritas? Potential limitations for SARS-CoV-2 vaccine development based on T-cell epitope prediction. PLoS Pathog. 2020, 16, e1008607. [Google Scholar] [CrossRef]
- Rivino, L.; Tan, A.T.; Chia, A.; Kumaran, E.A.P.; Grotenbreg, G.M.; MacAry, P.A.; Bertoletti, A. Defining CD8 + T Cell Determinants during Human Viral Infection in Populations of Asian Ethnicity. J. Immunol. 2013, 191, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Carragher, D.M.; Kaminski, D.A.; Moquin, A.; Hartson, L.; Randall, T.D. A Novel Role for Non-Neutralizing Antibodies against Nucleoprotein in Facilitating Resistance to Influenza Virus. J. Immunol. 2008, 181, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- Cardinaud, S.; Consiglieri, G.; Bouziat, R.; Urrutia, A.; Graff-Dubois, S.; Fourati, S.; Malet, I.; Guergnon, J.; Guihot, A.; Katlama, C.; et al. CTL escape mediated by proteasomal destruction of an HIV-1 cryptic epitope. PLoS Pathog. 2011, 7, e1002049. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Carlson, J.; Yan, J.; Akinsiku, O.T.; Schaefer, M.; Sabbaj, S.; Bet, A.; Levy, D.N.; Heath, S.; Tang, J.; et al. CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J. Exp. Med. 2010, 207, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.L.; Green, K.A.; Green, W.R. Alternative translational reading frames as a novel source of epitopes for an expanded CD8 T-cell repertoire: Use of a retroviral system to assess the translational requirements for CTL recognition and lysis. Viral Immunol. 2010, 23, 577–583. [Google Scholar] [CrossRef]
- Berger, C.T.; Carlson, J.M.; Brumme, C.J.; Hartman, K.L.; Brumme, Z.L.; Henry, L.M.; Rosato, P.C.; Piechocka-Trocha, A.; Brockman, M.A.; Harrigan, P.R.; et al. Viral adaptation to immune selection pressure by HLA class I-restricted CTL responses targeting epitopes in HIV frameshift sequences. J. Exp. Med. 2010, 207, 61–75. [Google Scholar] [CrossRef]
- Altfeld, M.; Addo, M.M.; Shankarappa, R.; Lee, P.K.; Allen, T.M.; Yu, X.G.; Rathod, A.; Harlow, J.; O’Sullivan, K.; Johnston, M.N.; et al. Enhanced Detection of Human Immunodeficiency Virus Type 1-Specific T-Cell Responses to Highly Variable Regions by Using Peptides Based on Autologous Virus Sequences. J. Virol. 2003, 77, 7330–7340. [Google Scholar] [CrossRef]
- Nickle, D.C.; Rolland, M.; Jensen, M.A.; Kosakovsky Pond, S.L.; Deng, W.; Seligman, M.; Heckerman, D.; Mullins, J.I.; Jojic, N. Coping with viral diversity in HIV vaccine design. PLoS Comput. Biol. 2007, 3, 754–762. [Google Scholar] [CrossRef]
- Rolland, M.; Manocheewa, S.; Swain, J.V.; Lanxon-Cookson, E.C.; Kim, M.; Westfall, D.H.; Larsen, B.B.; Gilbert, P.B.; Mullins, J.I. HIV-1 Conserved-Element Vaccines: Relationship between Sequence Conservation and Replicative Capacity. J. Virol. 2013, 87, 5461–5467. [Google Scholar] [CrossRef]
- Malhotra, U.; Nolin, J.; Mullins, J.I.; McElrath, M.J. Comprehensive epitope analysis of cross-clade Gag-specific T-cell responses in individuals with early HIV-1 infection in the US epidemic. Vaccine 2007, 25, 381–390. [Google Scholar] [CrossRef]
- Kesturu, G.S.; Colleton, B.A.; Liu, Y.; Heath, L.; Shaikh, O.S.; Rinaldo, C.R.; Shankarappa, R. Minimization of genetic distances by the consensus, ancestral, and center-of-tree (COT) sequences for HIV-1 variants within an infected individual and the design of reagents to test immune reactivity. Virology 2006, 348, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Jensen, M.A.; Nickle, D.C.; Yan, J.; Learn, G.H.; Heath, L.; Weiner, D.; Mullins, J.I. Reconstruction and Function of Ancestral Center-of-Tree Human Immunodeficiency Virus Type 1 Proteins. J. Virol. 2007, 81, 8507–8514. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.A.; Nickle, D.C.; Liu, Y.; Heath, L.; Jensen, M.A.; Rodrigo, A.G.; Mullins, J.I. Sources of variation in ancestral sequence reconstruction for HIV-1 envelope genes. Evol. Bioinform. Online 2007, 2, 53–76. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Gough, E.; Ritter, D.; Wilson, C.; Mulenga, J.; Allen, S.; Goepfert, P.A. Group M-based HIV-1 Gag peptides are frequently targeted by T cells in chronically infected US and Zambian patients. AIDS 2006, 20, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Posada, D. Computational Design of Centralized HIV-1 Genes. Curr. HIV Res. 2011, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Kothe, D.L.; Li, Y.; Decker, J.M.; Bibollet-Ruche, F.; Zammit, K.P.; Salazar, M.G.; Chen, Y.; Weng, Z.; Weaver, E.A.; Gao, F.; et al. Ancestral and consensus envelope immunogens for HIV-1 subtype C. Virology 2006, 352, 438–449. [Google Scholar] [CrossRef]
- Rutebemberwa, A.; Currier, J.R.; Jagodzinski, L.; McCutchan, F.; Birx, D.; Marovich, M.; Cox, J.H. HIV-1 MN Env 15-mer peptides better detect HIV-1 specific CD8 T cell responses compared with consensus subtypes B and M group 15-mer peptides. AIDS 2005, 19, 1165–1172. [Google Scholar] [CrossRef]
- De Groot, A.S.; Bishop, E.A.; Khan, B.; Lally, M.; Marcon, L.; Franco, J.; Mayer, K.H.; Carpenter, C.C.J.; Martin, W. Engineering immunogenic consensus T helper epitopes for a cross-clade HIV vaccine. Methods 2004, 34, 476–487. [Google Scholar] [CrossRef]
- Koita, O.A.; Dabitao, D.; Mahamadou, I.; Tall, M.; Dao, S.; Tounkara, A.; Guiteye, H.; Noumsi, C.; Thiero, O.; Kone, M.; et al. Confirmation of immunogenic consensus sequence HIV-1 T-cell epitopes in Bamako, Mali and Providence, Rhode Island. Hum. Vaccin. 2006, 2, 119–128. [Google Scholar] [CrossRef]
- Almeida, R.R.; Rosa, D.S.; Ribeiro, S.P.; Santana, V.C.; Kallás, E.G.; Sidney, J.; Sette, A.; Kalil, J.; Cunha-Neto, E. Broad and Cross-Clade CD4+ T-Cell Responses Elicited by a DNA Vaccine Encoding Highly Conserved and Promiscuous HIV-1 M-Group Consensus Peptides. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Coutinho-Silva, A.; Fonseca, L.A.M.; Segurado, A.C.; Moraes, S.L.; Rodrigues, H.; Hammer, J.; Kallás, E.G.; Sidney, J.; Sette, A.; et al. Identification of novel consensus CD4 T-cell epitopes from clade B HIV-1 whole genome that are frequently recognized by HIV-1 infected patients. AIDS 2006, 20, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Frahm, N.; Nickle, D.C.; Linde, C.H.; Cohen, D.E.; Zuniga, R.; Lucchetti, A.; Roach, T.; Walker, B.D.; Allen, T.M.; Korber, B.T.; et al. Increased detection of HIV-specific T cell responses by combination of central sequences with comparable immunogenicity. AIDS 2008, 22, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Brander, C.; Self, S.; Korber, B. Capturing viral diversity for in-vitro test reagents and HIV vaccine immunogen design. Curr. Opin. HIV AIDS 2007, 2, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Melamed, S.; Weiss, S.; Israely, T.; et al. The coding capacity of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- PeptGen Peptide Generator. Available online: https://www.hiv.lanl.gov/content/sequence/PEPTGEN/peptgen.html (accessed on 3 August 2020).
- Llano, A.; Cedeño, S.; Silva Arrieta, S.; Brander, C.; Theoretical Biology and Biophysics Group. The 2019 Optimal HIV CTL epitopes update: Growing diversity in epitope length and HLA restriction. In HIV Molecular Immunology; Yusim, K., Korber, B., Brander, C., Barouch, D., de Boer, R., Haynes, B.F., Koup, R., Moore, J.P., Walker, B., Eds.; Los Alamos National Laboratory: Los Alamos, NM, USA, 2019. [Google Scholar]
- Wagih, O. Ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 2017, 33, 3645–3647. [Google Scholar] [CrossRef]
- BLAST: Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 3 August 2020).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- WebLogo. Available online: http://weblogo.berkeley.edu/ (accessed on 3 August 2020).
- IEDB.org: Free Epitope Database and Prediction Resource. Available online: http://www.iedb.org/ (accessed on 3 August 2020).
- Draenert, R.; Altfeld, M.; Brander, C.; Basgoz, N.; Corcoran, C.; Wurcel, A.G.; Stone, D.R.; Kalams, S.A.; Trocha, A.; Addo, M.M.; et al. Comparison of overlapping peptide sets for detection of antiviral CD8 and CD4 T cell responses. J. Immunol. Methods 2003, 275, 19–29. [Google Scholar] [CrossRef]
- Frahm, N.; Korber, B.T.; Adams, C.M.; Szinger, J.J.; Draenert, R.; Addo, M.M.; Feeney, M.E.; Yusim, K.; Sango, K.; Brown, N.V.; et al. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J. Virol. 2004, 78, 2187–2200. [Google Scholar] [CrossRef] [PubMed]
- Yerly, D.; Heckerman, D.; Allen, T.M.; Chisholm, J.V.; Faircloth, K.; Linde, C.H.; Frahm, N.; Timm, J.; Pichler, W.J.; Cerny, A.; et al. Increased Cytotoxic T-Lymphocyte Epitope Variant Cross-Recognition and Functional Avidity Are Associated with Hepatitis C Virus Clearance. J. Virol. 2008, 82, 3147–3153. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. Presence of SARS-CoV-2 reactive T cells in COVID-19 patients and healthy donors. medRxiv 2020. [Google Scholar] [CrossRef]
- Prado, J.G.; Honeyborne, I.; Brierley, I.; Puertas, M.C.; Martinez-Picado, J.; Goulder, P.J.R. Functional Consequences of Human Immunodeficiency Virus Escape from an HLA-B*13-Restricted CD8+ T-Cell Epitope in p1 Gag Protein. J. Virol. 2009, 83, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Honeyborne, I.; Codoñer, F.M.; Leslie, A.; Tudor-Williams, G.; Luzzi, G.; Ndung’u, T.; Walker, B.D.; Goulder, P.J.; Prado, J.G. HLA-Cw*03-Restricted CD8+ T-Cell Responses Targeting the HIV-1 Gag Major Homology Region Drive Virus Immune Escape and Fitness Constraints Compensated for by Intracodon Variation. J. Virol. 2010, 84, 11279–11288. [Google Scholar] [CrossRef]
- De Campos-Lima, P.O.; Gavioli, R.; Zhang, Q.J.; Wallace, L.E.; Dolcetti, R.; Rowe, M.; Rickinson, A.B.; Masucci, M.G. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11 + population. Science 1993, 260, 98–100. [Google Scholar] [CrossRef]
- Gutiérrez, M.I.; Spangler, G.; Kingma, D.; Raffeld, M.; Guerrero, I.; Misad, O.; Jaffe, E.S.; Magrath, I.T.; Bhatia, K. Epstein-Barr virus in nasal lymphomas contains multiple ongoing mutations in the EBNA-1 gene. Blood 1998, 92, 600–606. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Honnayakanahalli Marichann, M.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through Recombination and Strong Purifying Selection Short Title: Recombination and origin of SARS-CoV-2 One Sentence Summary: Extensive Recombination and Strong Purifying Selection among coronaviruses from different hosts facilita. bioRxiv 2020. [Google Scholar] [CrossRef]
- Frahm, N.; Kaufmann, D.E.; Yusim, K.; Muldoon, M.; Kesmir, C.; Linde, C.H.; Fischer, W.; Allen, T.M.; Li, B.; McMahon, B.H.; et al. Increased sequence diversity coverage improves detection of HIV-specific T cell responses. J Immunol 2007, 179, 6638–6650. [Google Scholar] [CrossRef]
- Ng, K.; Faulkner, N.; Cornish, G.; Rosa, A.; Earl, C.; Wrobel, A.; Benton, D.; Roustan, C.; Bolland, W.; Thompson, R.; et al. Pre-existing and de novo humoral immunity to SARS-CoV-2 in humans. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lascano, A.M.; Epiney, J.B.; Coen, M.; Serratrice, J.; Bernard-Valnet, R.; Lalive, P.H.; Kuntzer, T.; Hübers, A. SARS-CoV-2 and Guillain-Barré syndrome: AIDP variant with favorable outcome. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bigaut, K.; Mallaret, M.; Baloglu, S.; Nemoz, B.; Morand, P.; Baicry, F.; Godon, A.; Voulleminot, P.; Kremer, L.; Chanson, J.-B.; et al. Guillain-Barré syndrome related to SARS-CoV-2 infection. Neurol. Neuroimmunol. Neuroinflammation 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Russo, T.; Falzone, Y.M.; Strollo, M.; Amadio, S.; Del Carro, U.; Locatelli, M.; Filippi, M.; Fazio, R. Post-infectious Guillain-Barré syndrome related to SARS-CoV-2 infection: A case report. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Ebadi, H.; Sarna, J.R. Guillain-Barré syndrome with facial diplegia related to SARS-CoV-2 infection. Can. J. Neurol. Sci. 2020, 1–10. [Google Scholar] [CrossRef]
- Draenert, R.; Brander, C.; Yu, X.G.; Altfeld, M.; Verrill, C.L.; Feeney, M.E.; Walker, B.D.; Goulder, P.J.R. Impact of intrapeptide epitope location on CD8 T cell recognition: Implications for design of overlapping peptide panels. AIDS 2004, 18, 871–876. [Google Scholar] [CrossRef]



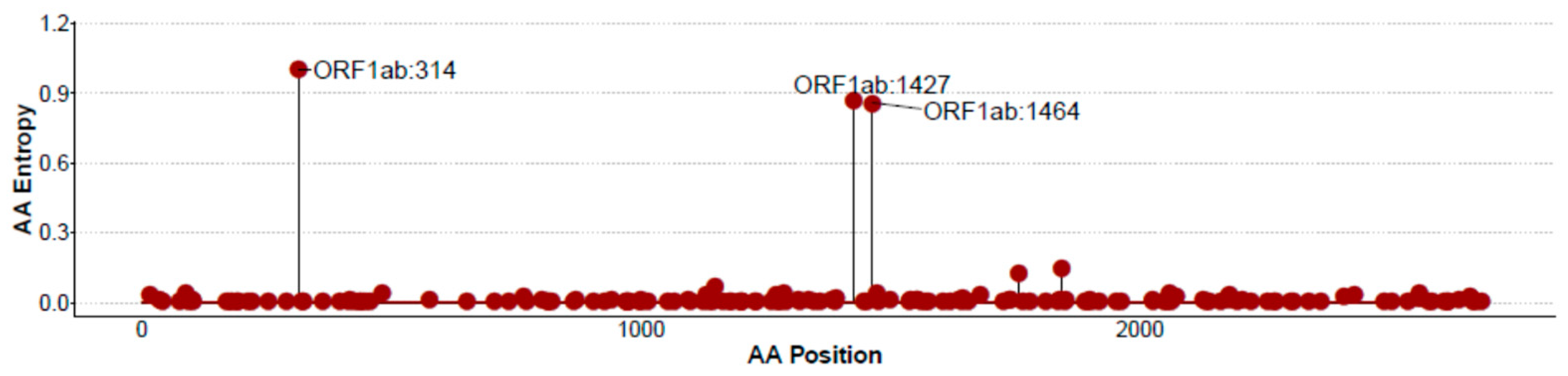{kind=link}
{kind=link}
| Gene | Start | End | Protein | Protease Products | Frame |
|---|---|---|---|---|---|
| ORF1a.iORF1.ext | 59 | 136 | upORF1a1 | - | Alternative |
| ORF1a.iORF2.ext | 163 | 264 | upORF1a2 | - | Alternative |
| ORF1ab | 266 | 13483 | pp1a | leader protein | Canonical |
| nsp2 | |||||
| nsp3 | |||||
| nsp4 | |||||
| 3C-like proteinase | |||||
| nsp6 | |||||
| nsp7 | |||||
| nsp8 | |||||
| nsp9 | |||||
| nsp10 | |||||
| nsp11 | |||||
| ORF1ab | 13468 | 21555 | pp1ab | RNA-dependent RNA polymerase | Canonical |
| helicase | |||||
| 3′-to-5′ exonuclease | |||||
| endoRNAse | |||||
| 2′-O-ribose methyltransferase | |||||
| S | 21563 | 25384 | surface glycoprotein | S1 | Canonical |
| S2 | |||||
| ORFS.iORF1 | 21744 | 21863 | inORFS | - | Alternative |
| ORF3a | 25393 | 26220 | ORF3a protein | - | Canonical |
| ORF3a.iORF1 | 25457 | 25582 | inORF3a1 | - | Alternative |
| ORF3a.iORF2 | 25596 | 25697 | inORF3a2 | - | Alternative |
| E | 26245 | 26472 | envelope protein | - | Canonical |
| ORFM.ext | 26484 | 27191 | exORFM | - | Alternative |
| M | 26523 | 27191 | membrane glycoprotein | - | Canonical |
| ORFM.iORF | 27151 | 27195 | inORFM | - | Alternative |
| ORF6 | 27202 | 27387 | ORF6 protein | - | Canonical |
| ORF7a | 27394 | 27759 | ORF7a protein | - | Canonical |
| ORF7b | 27756 | 27887 | ORF7b protein | - | Canonical |
| ORF7b.iORF2 | 27862 | 27897 | inORF7b | - | Alternative |
| ORF8 | 27894 | 28259 | ORF8 protein | - | Canonical |
| ORF8.iORF | 27965 | 27994 | inORF8 | - | Alternative |
| N | 28274 | 29533 | nucleocapsid phosphoprotein | - | Canonical |
| ORFN.iORF1 | 28284 | 28577 | ORF9b | - | Alternative |
| ORF10.upORF | 29538 | 29570 | upORF10 | - | Alternative |
| ORF10 | 29558 | 29674 | ORF10 protein | - | Canonical |
| Set | Length | Overlapp | Number | Variants |
|---|---|---|---|---|
| 15–11 | 15 | 11 | 2821 | 31 |
| 15–10 | 15 | 10 | 2262 | 23 |
| 18–11 | 18 | 11 | 1561 | 22 |
| Consensus Sequence | ORF | Consensus Start Position | Alignment Hit | Epitopes | |||||
|---|---|---|---|---|---|---|---|---|---|
| I | II | III | Unknown | SARS-CoV | Human | Other Coronavirus | |||
| VGVLTLDNQDLNG | ORF1b | 193 | 1 | 4 | - | - | |||
| TQMNLKYAISAKNRARTVAGVSI | ORF1b | 530 | - | 5 | 2 | - | |||
| VIGTSKFYGGW | ORF1b | 580 | - | 3 | - | - | |||
| LMGWDYPKCDRAMPN | ORF1b | 605 | 1 | 3 | - | - | |||
| LANECAQVL | ORF1b | 646 | - | 1 | - | - | |||
| YVKPGGTSSGDATTA | ORF1b | 665 | - | 3 | - | - | |||
| KHFSMMILSDDAVVCFN | ORF1b | 743 | - | 2 | 1 | - | |||
| LYYQNNVFMS | ORF1b | 778 | - | - | - | - | |||
| GPHEFCSQHT | ORF1b | 800 | - | 2 | - | - | |||
| LPYPDPSRIL | ORF1b | 820 | - | 2 | 3 | - | |||
| ERFVSLAIDAYPL | ORF1b | 849 | - | 5 | - | 1 | |||
| SQTSLRCG | ORF1b | 934 | - | 1 | - | - | |||
| LYLGGMSYY | ORF1b | 986 | - | 3 | - | - | |||
| LKLFAAET | ORF1b | 1054 | - | 4 | - | - | |||
| QGPPGTGKSH | ORF1b | 1205 | 1 | 2 | 40 | - | |||
| TACSHAAVDALCEKA | ORF1b | 1231 | - | 1 | - | - | |||
| GDPAQLPAPR | ORF1b | 1324 | - | 3 | - | - | |||
| AVFISPYNSQN | ORF1b | 1432 | - | 4 | 1 | - | |||
| NRFNVAITRA | ORF1b | 1483 | - | 2 | - | - | |||
| CNLGGAVC | ORF1b | 2002 | - | 1 | - | - | |||
| KYTQLCQYLN | ORF1b | 2443 | - | 3 | - | - | |||
| RSFIEDLLF | Spike | 815 | - | 2 | - | - | |||
| QIDRLITGRL | Spike | 993 | - | 5 | - | 1 | |||
| KWPWYIWL | Spike | 1211 | - | - | - | - | |||
| WSFNPETN | M | 110 | - | 3 | - | - | |||
| PRWYFYYLGTGP | N | 106 | - | 7 | - | - | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olvera, A.; Noguera-Julian, M.; Kilpelainen, A.; Romero-Martín, L.; Prado, J.G.; Brander, C. SARS-CoV-2 Consensus-Sequence and Matching Overlapping Peptides Design for COVID19 Immune Studies and Vaccine Development. Vaccines 2020, 8, 444. https://doi.org/10.3390/vaccines8030444
Olvera A, Noguera-Julian M, Kilpelainen A, Romero-Martín L, Prado JG, Brander C. SARS-CoV-2 Consensus-Sequence and Matching Overlapping Peptides Design for COVID19 Immune Studies and Vaccine Development. Vaccines. 2020; 8(3):444. https://doi.org/10.3390/vaccines8030444
Chicago/Turabian StyleOlvera, Alex, Marc Noguera-Julian, Athina Kilpelainen, Luis Romero-Martín, Julia G. Prado, and Christian Brander. 2020. "SARS-CoV-2 Consensus-Sequence and Matching Overlapping Peptides Design for COVID19 Immune Studies and Vaccine Development" Vaccines 8, no. 3: 444. https://doi.org/10.3390/vaccines8030444
APA StyleOlvera, A., Noguera-Julian, M., Kilpelainen, A., Romero-Martín, L., Prado, J. G., & Brander, C. (2020). SARS-CoV-2 Consensus-Sequence and Matching Overlapping Peptides Design for COVID19 Immune Studies and Vaccine Development. Vaccines, 8(3), 444. https://doi.org/10.3390/vaccines8030444