
A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques


Abstract
:1. Introduction
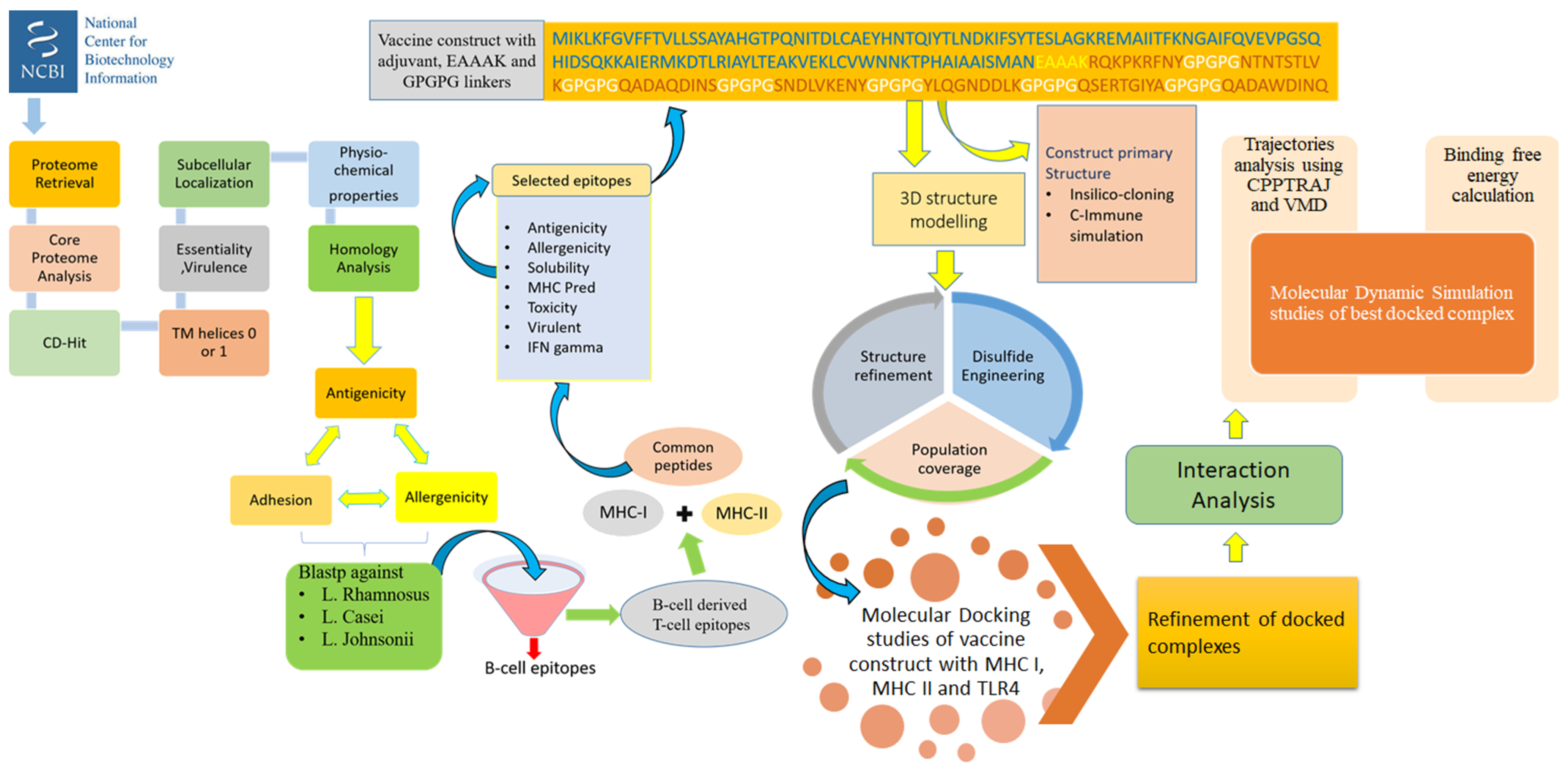
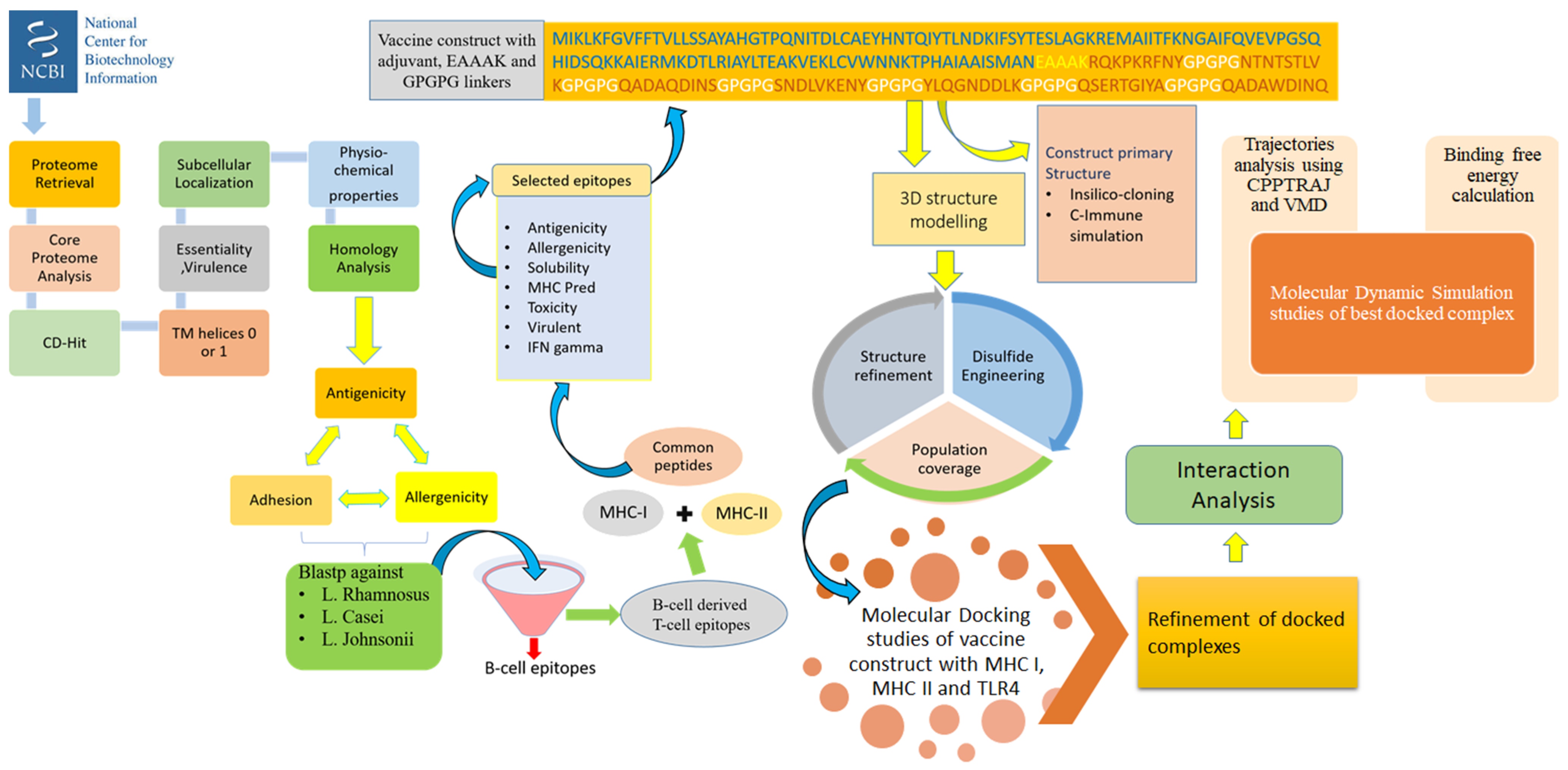
2. Methodology
2.1. Proteome Retrieval and Subcellular Localization
2.2. Vaccine Candidate Prioritization Phase
2.3. B-Cell and T-Cell Epitopes Prediction
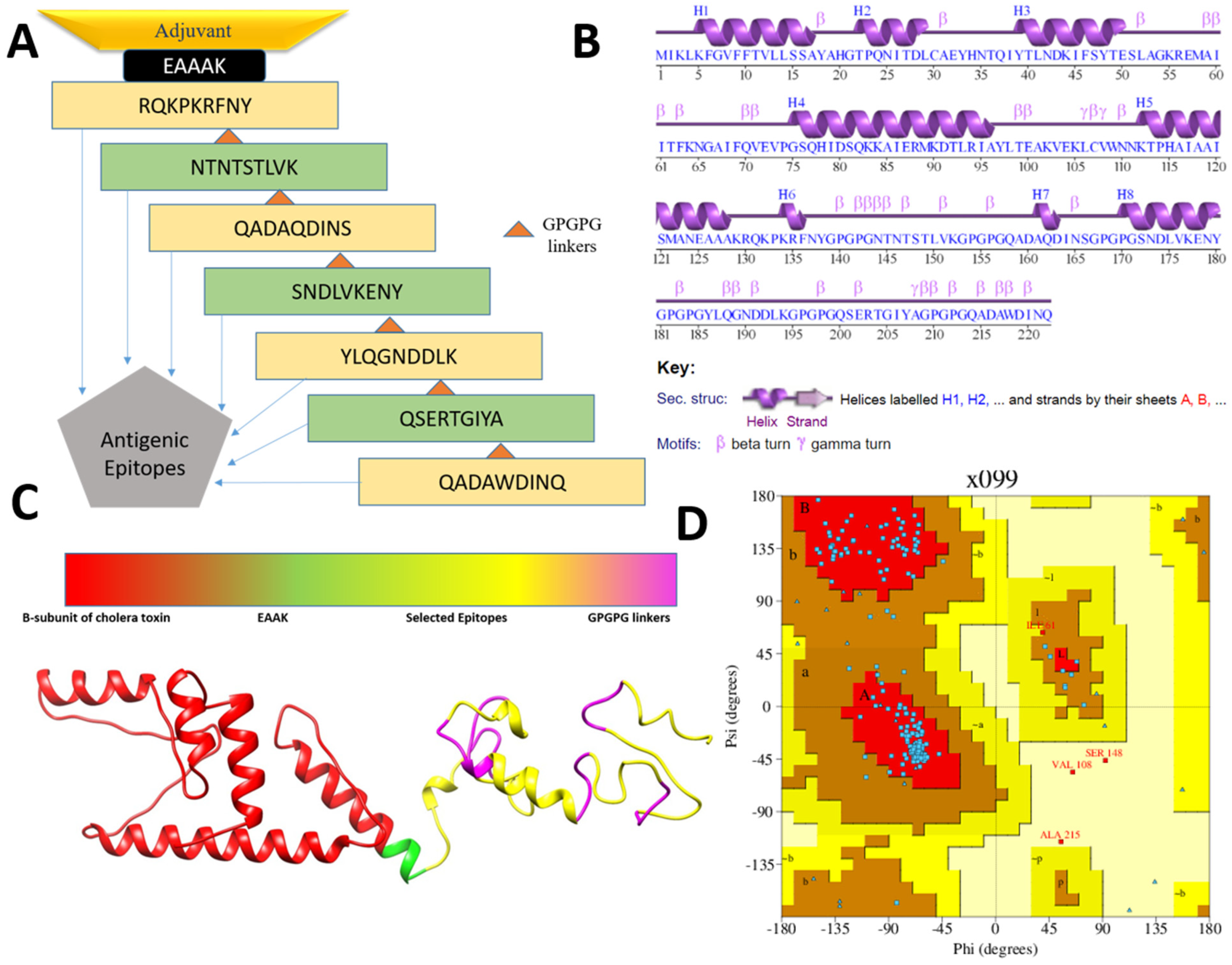
2.4. Designing and Processing of Vaccine Construct
2.5. Blind Docking Analysis
2.6. Molecular Dynamics Simulation Assay
2.7. Estimation of Binding Free Energy
2.8. In Silico Immune Profiling of Multi-Epitopes Vaccine Construct
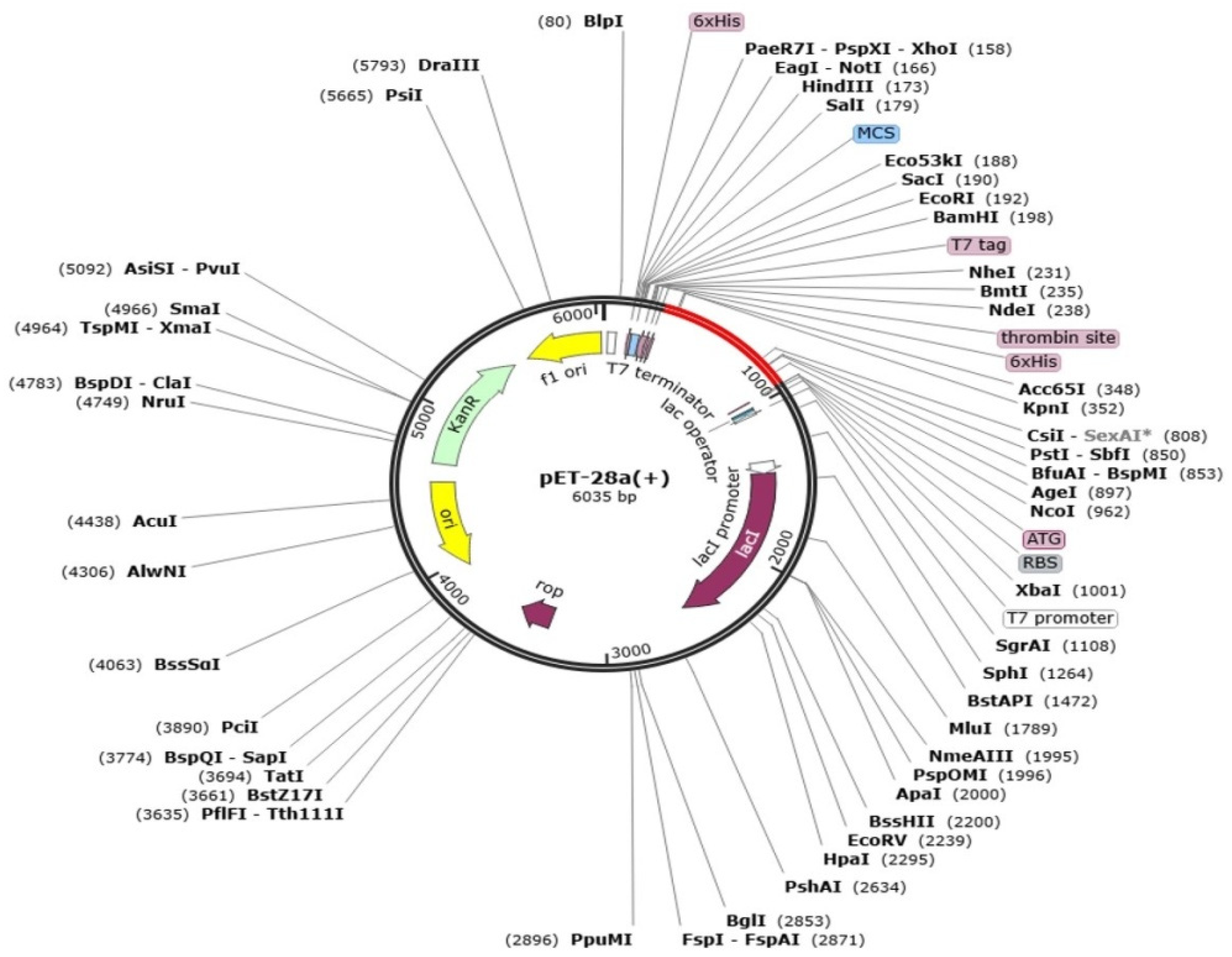
2.9. Codon Adaptation and Cloning
2.10. Disulfide Engineering
3. Results and Discussion
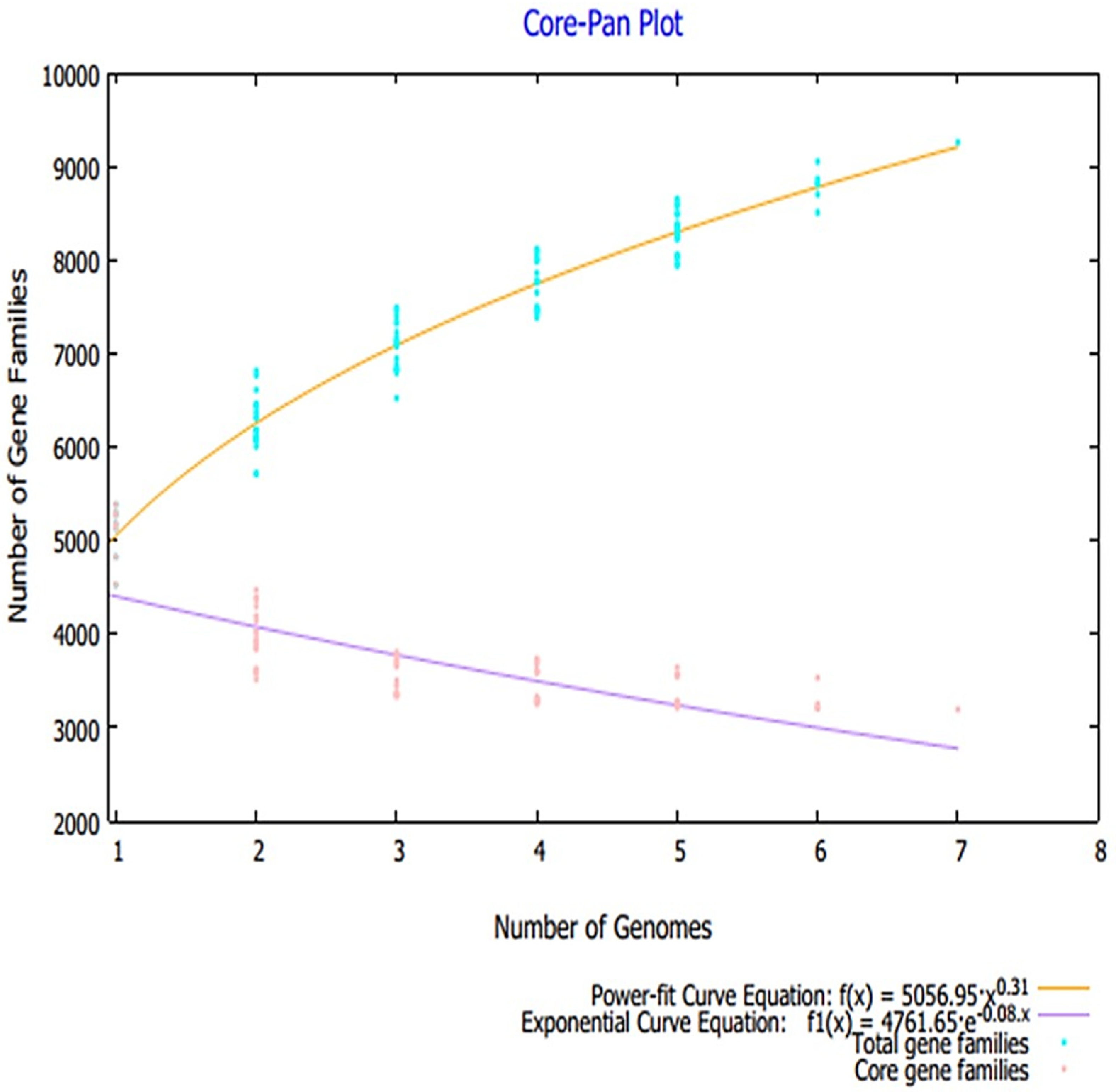
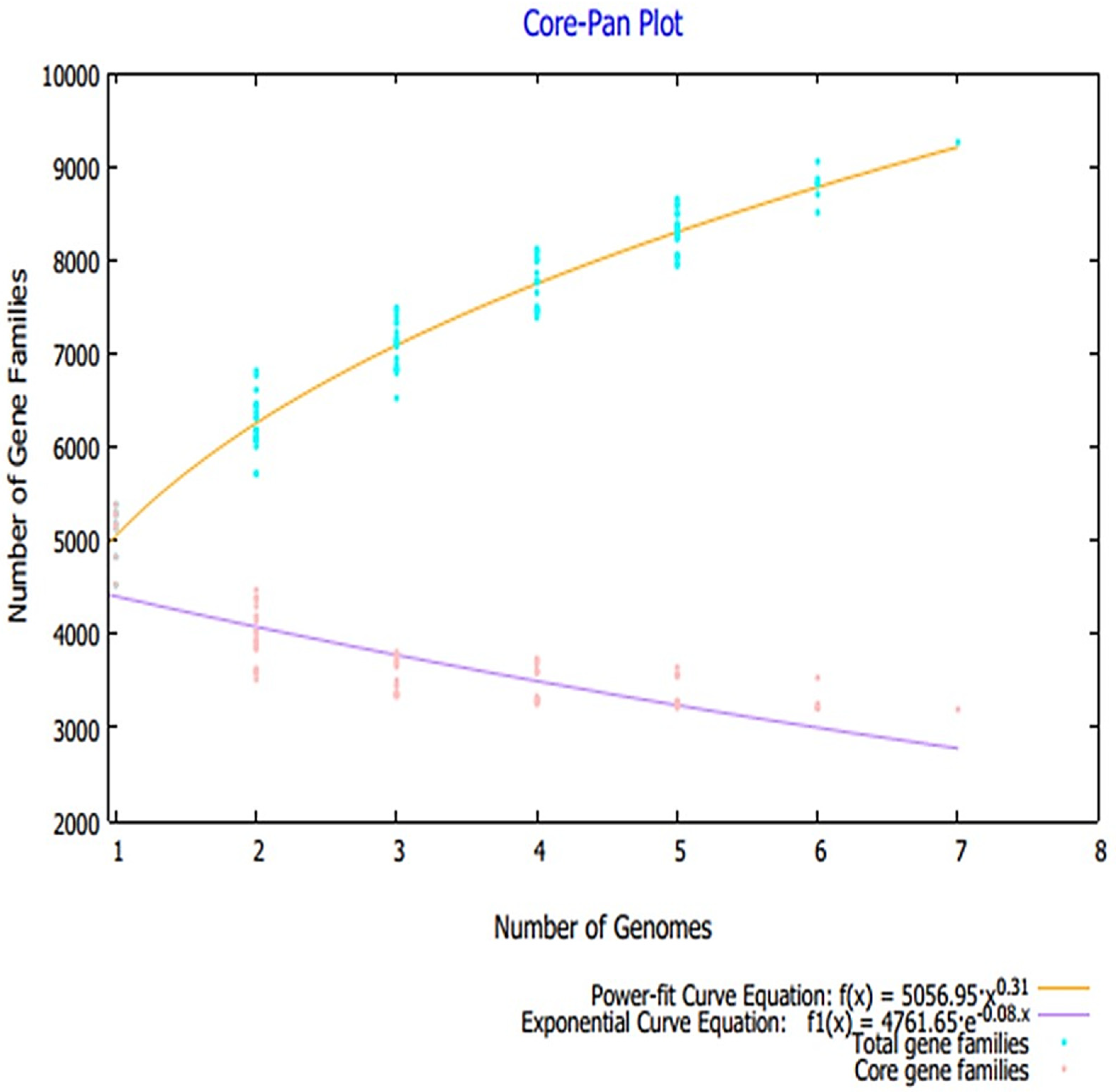
3.1. Pan-Proteome Analysis and Prioritization of Potential Vaccine Candidates
3.2. Mapping of B and T Cell Epitopes
3.3. Multi-Epitopes Vaccine Construct
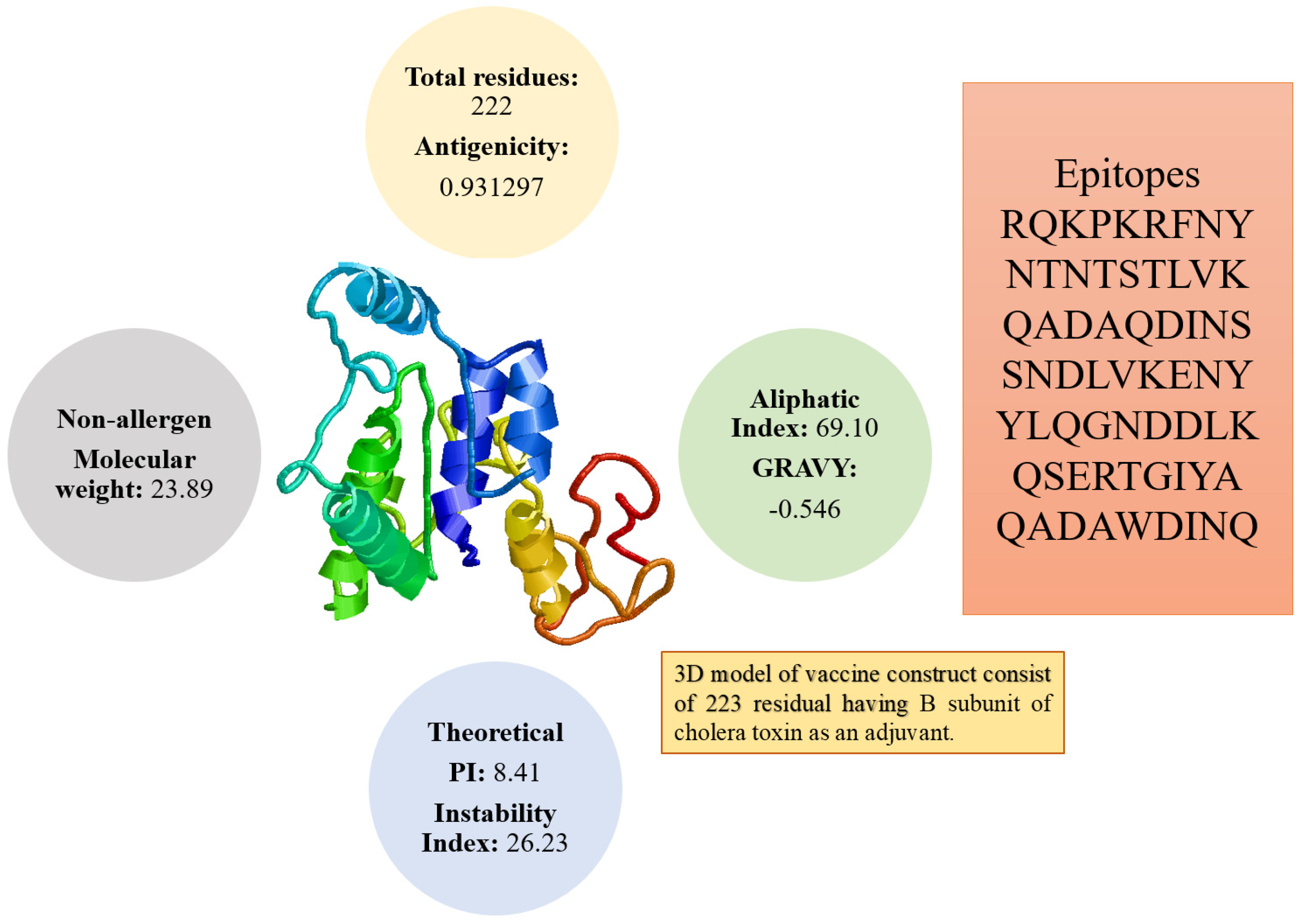
3.4. Three Dimensional Structure Modeling and Processing
3.5. Physiochemical Analysis of the Vaccine
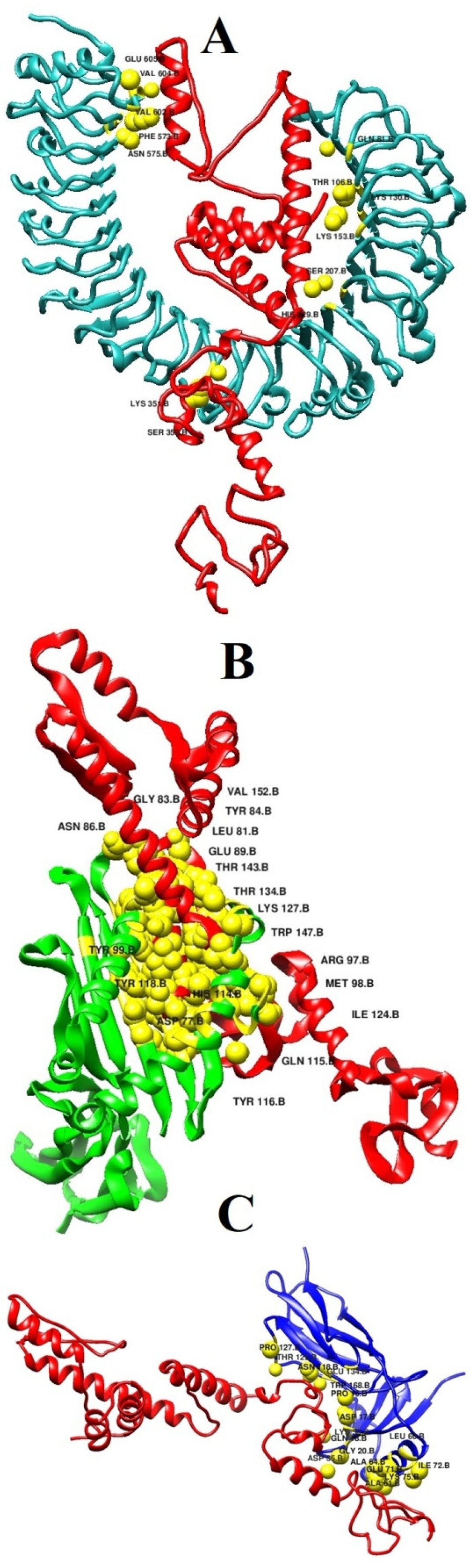
3.6. Interaction Analysis
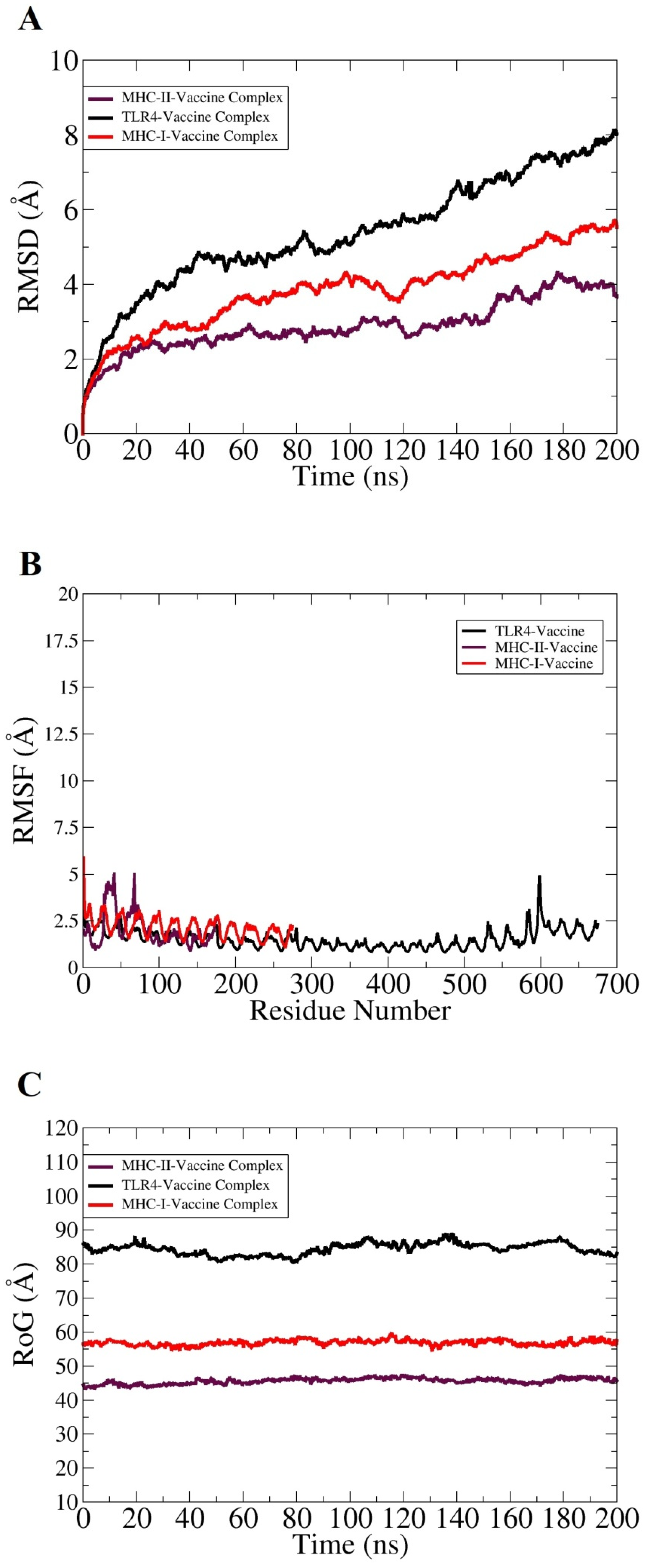
3.7. Molecular Dynamics Simulation Analysis
3.8. Calculation of Binding Free Energies
3.9. In Silico Expression Analysis of Multi-Epitope Vaccine Construct
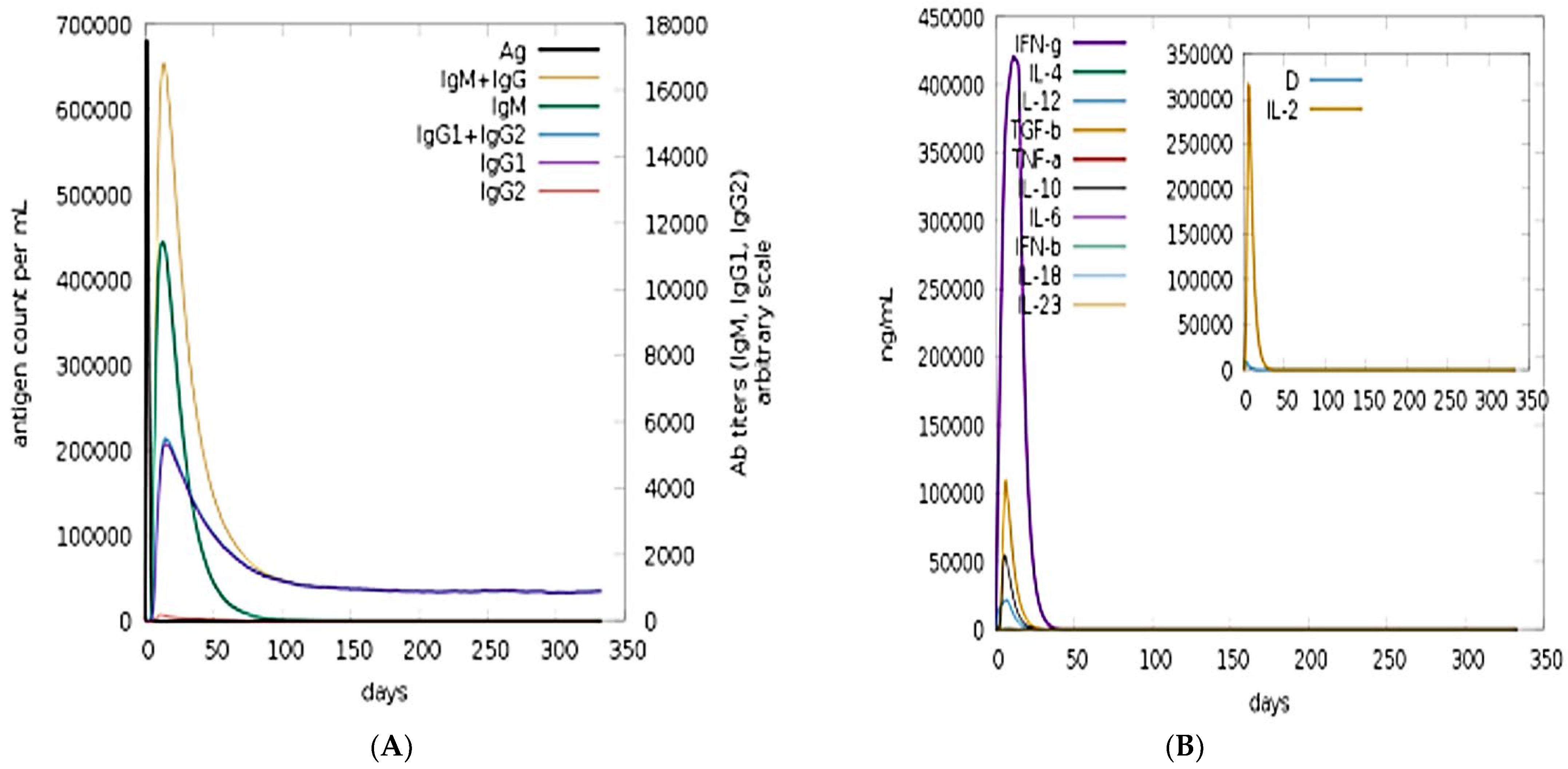
3.10. Immune Simulation
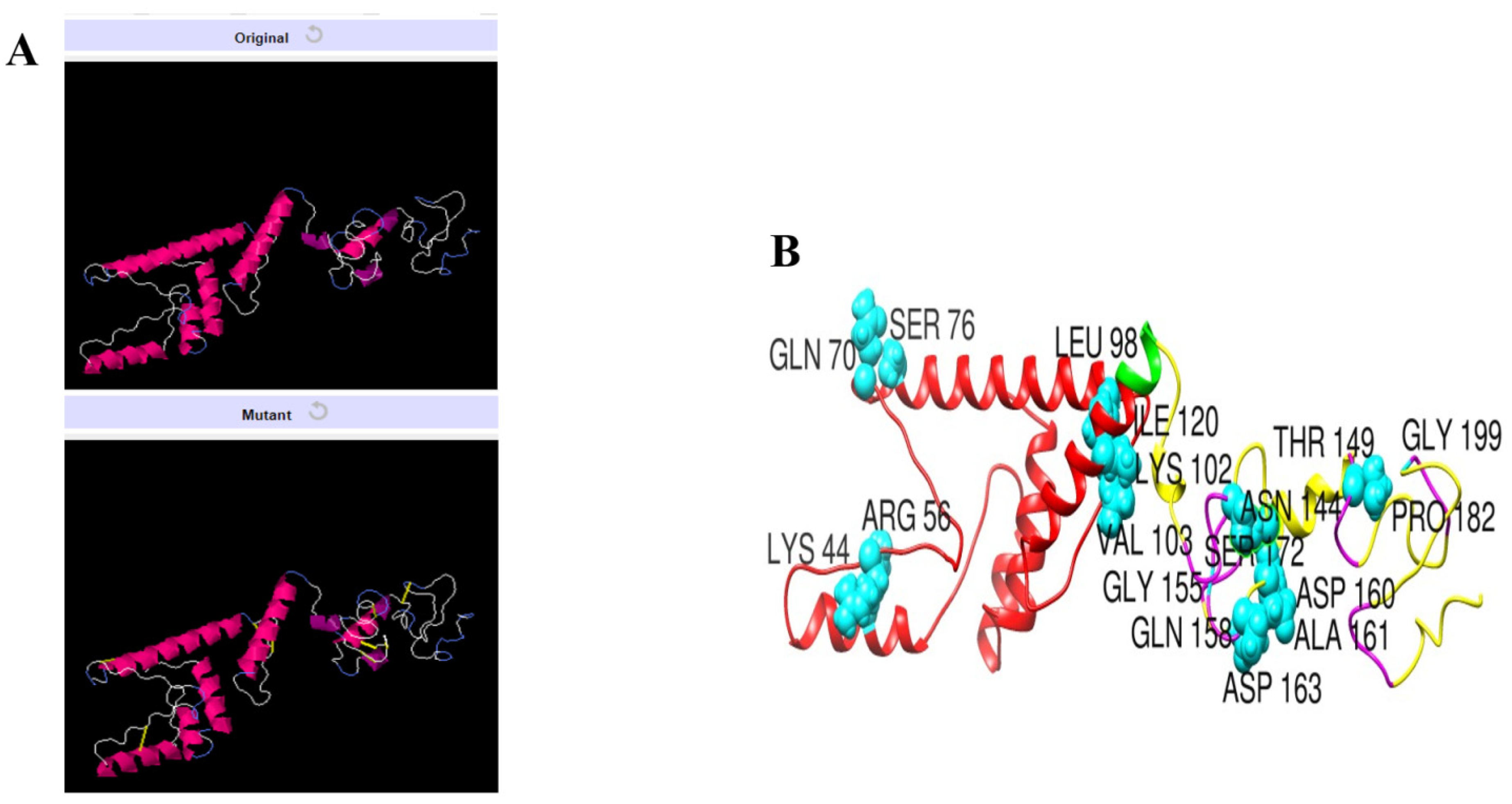
3.11. Disulfide Engineering of the Vaccine
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, M.; Li, X. Klebsiella pneumoniae and Pseudomonas aeruginosa. In Molecular Medical Microbiology; Elsevier BV: Amsterdam, The Netherlands, 2015; pp. 1547–1564. [Google Scholar]
- Rathi, C.R.; Suresh, S.N.; Geethalakshmi, S.; Ilangovan, M.; Rasheed, R.; Vazhacharickal, P.J. Synthesis of Silver Nanoparticles from Mirabilis Jalapa and Evaluation of Antioxidant Activity; Prem Jose, Independently published; Available online: https://books.google.co.id/books?hl=zh-CN&lr=&id=5FIOEAAAQBAJ&oi=fnd&pg=PA19&dq=related:ritE0Q9u6C0J:scholar.google.com/&ots=zLYAn-HrW0&sig=Z6rhE7xILQbzOlJDgnI4u-ziVtA&redir_esc=y#v=onepage&q&f=false (accessed on 11 December 2020).
- Ryan, K.J.; Ray, C.G. Medical Microbiology; McGraw Hill: New York, NY, USA, 2004; Volume 4, p. 370. [Google Scholar]
- Podschun, R.; Ullmann, U. Klebsiella spp. as Nosocomial Pathogens: Epidemiology, Taxonomy, Typing Methods, and Pathogenicity Factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagt, E.W.; Short, S. Healthcare-Associated Infections. In Pediatric Critical Care; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1105–1143. [Google Scholar]
- Rashid, T.; Ebringer, A.; Wilson, C. The role of Klebsiella in Crohn’s disease with a potential for the use of antimicrobial measures. Int. J. Rheumatol. 2013, 2013, 610393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, P.Y. The emerging problems of Klebsiella pneumoniae infections: Carbapenem resistance and biofilm formation. FEMS Microbiol. Lett. 2016, 363, fnw219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cillóniz, C.; Dominedò, C.; Torres, A. Multidrug resistant gram-negative bacteria in community-acquired pneumonia. Annu. Update Intensiv. Care Emerg. Med. 2019, 2019, 459–475. [Google Scholar]
- Ribeiro, S.; De La Fuente-Núñez, C.; Baquir, B.; Faria-Junior, C.; Franco, O.L.; Hancock, R. Antibiofilm Peptides Increase the Susceptibility of Carbapenemase-Producing Klebsiella pneumoniae Clinical Isolates to β-Lactam Antibiotics. Antimicrob. Agents Chemother. 2015, 59, 3906–3912. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, U.; Aggarwal, R. Extended spectrum β-lactamases (ESBL)—An emerging threat to clinical therapeutics. Indian J. Med. Microbiol. 2004, 22, 75–80. [Google Scholar] [CrossRef]
- Lee, C.-H.; Su, L.-H.; Tang, Y.-F.; Liu, J.-W. Treatment of ESBL-producing Klebsiella pneumoniae bacteraemia with carbapenems or flomoxef: A retrospective study and laboratory analysis of the isolates. J. Antimicrob. Chemother. 2006, 58, 1074–1077. [Google Scholar] [CrossRef] [Green Version]
- Rice, L.B. Mechanisms of Resistance and Clinical Relevance of Resistance to β-Lactams, Glycopeptides, and Fluoroquinolones. In Proceedings of the Mayo Clinic Proceedings; Elsevier BV: Amsterdam, The Netherlands, 2012; Volume 87, pp. 198–208. [Google Scholar]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-lactamase (NDM): A threat to public health. BMC Microbiol. 2017, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Fazeli, H.; Norouzi-Barough, M.; Ahadi, A.M.; Shokri, D.; Solgi, H. Detection of New Delhi Metallo-Beta-Lactamase-1 (NDM-1) in carbapenem- resistant Klebsiella pneumoniae isolated from a university hospital in Iran. Hippokratia 2016, 19, 205–209. [Google Scholar]
- Logan, L.K.; Weinstein, R.A. The Epidemiology of Carbapenem-Resistant Enterobacteriaceae: The Impact and Evolution of a Global Menace. J. Infect. Dis. 2017, 215, S28–S36. [Google Scholar] [CrossRef] [Green Version]
- Grundmann, H.; Livermore, D.M.; Giske, C.G.; Cantón, R.; Rossolini, G.M.; Campos, J.; Vatopoulos, A.; Gniadkowski, M.; Toth, A.; Pfeifer, Y.; et al. Carbapenem-non-susceptible Enterobacteriaceae in Europe: Conclusions from a meeting of national experts. Eurosurveillance 2010, 15, 19711. [Google Scholar] [CrossRef] [Green Version]
- Navarro, E.D.; Motley, M.P.; Pérez, G.R.; Yu, W.; Austin, J.; Seco, B.M.S.; Xiao, G.; Chikhalya, A.; Seeberger, P.H.; Fries, B.C. Erratum for Diago-Navarro et al. Novel, Broadly Reactive Anticapsular Antibodies against Carbapenem-Resistant Klebsiella pneumoniae Protect from Infection. mBio 2018, 9, e00091-18. [Google Scholar] [CrossRef] [Green Version]
- Nørgaard, S.M.; Jensen, C.S.; Aalestrup, J.; Vandenbroucke-Grauls, C.M.J.E.; De Boer, M.G.J.; Pedersen, A.B. Choice of therapeutic interventions and outcomes for the treatment of infections caused by multidrug-resistant gram-negative pathogens: A systematic review. Antimicrob. Resist. Infect. Control 2019, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Ah, Y.-M.; Kim, A.-J.; Lee, J.-Y. Colistin resistance in Klebsiella pneumoniae. Int. J. Antimicrob. Agents 2014, 44, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Tennant, S.M.; Simon, R.; Cross, A.S. Progress towards the development of Klebsiella vaccines. Expert Rev. Vaccines 2019, 18, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, C.D.; Telford, J.L.; Rappuoli, R.; Seib, K. Vaccinology in the genome era. J. Clin. Investig. 2009, 119, 2515–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Manzoor, K.N.; Sultan, A.; Saeed, M.; Rafique, M.; Noushad, S.; Talib, A.; Rentschler, S.; Deigner, H.-P. Pulling the Brakes on Fast and Furious Multiple Drug-Resistant (MDR) Bacteria. Int. J. Mol. Sci. 2021, 22, 859. [Google Scholar] [CrossRef]
- Naz, K.; Naz, A.; Ashraf, S.T.; Rizwan, M.; Ahmad, J.; Baumbach, J.; Ali, A. PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome. BMC Bioinform. 2019, 20, 123. [Google Scholar] [CrossRef]
- Delany, I.; Rappuoli, R.; Seib, K. Vaccines, Reverse Vaccinology, and Bacterial Pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a012476. [Google Scholar] [CrossRef] [Green Version]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; Qamar, M.T.U. Pan-vaccinomics approach towards a universal vaccine candidate against WHO priority pathogens to address growing global antibiotic resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ismail, S.; Ahmad, S.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-proteome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Wagner, J.R.; Laird, M.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.; Aljasir, M.; Alhumeed, N.; Qasim, M.; Ashfaq, U.; Qamar, M.T.U. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baseer, S.; Ahmad, S.; Ranaghan, K.; Azam, S.S. Towards a peptide-based vaccine against Shigella sonnei: A subtractive reverse vaccinology based approach. Biology 2017, 50, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, M.T.U.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K.; Tiwari, S.; Jamal, S.B.; Barh, D.; Azevedo, V.; Soares, S.C. An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. Int. J. Mol. Sci. 2017, 18, 402. [Google Scholar] [CrossRef]
- Ahmad, S.; Shahid, F.; Qamar, M.T.U.; Rehman, H.; Abbasi, S.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.; Almatroudi, A.; et al. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- ExPASy ProtParam. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 3 August 2021).
- Ahmad, S.; Navid, A.; Farid, R.; Abbas, G.; Ahmad, F.; Zaman, N.; Parvaiz, N.; Azam, S.S. Design of a Novel Multi Epitope-Based Vaccine for Pandemic Coronavirus Disease (COVID-19) by Vaccinomics and Probable Prevention Strategy against Avenging Zoonotics. Eur. J. Pharm. Sci. 2020, 151, 105387. [Google Scholar] [CrossRef]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [Green Version]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, G.; Kumar, K.; Jain, P.; Ramachandran, S. SPAAN: A software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics 2004, 21, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Mahajan, S.; Overton, J.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Guan, P.; Doytchinova, I.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. Peptide Toxicity Prediction. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2014; Volume 1268, pp. 143–157. [Google Scholar]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijker, M.S.; Melief, C.J.M.; Offringa, R.; Van Der Burg, S.H. Design and development of synthetic peptide vaccines: Past, present and future. Expert Rev. Vaccines 2007, 6, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Reche, P.A.; Fernandez-Caldas, E.; Flower, D.R.; Fridkis-Hareli, M.; Hoshino, Y. Peptide-based immunotherapeutics and vaccines. J. Immunol. Res. 2014, 2014, 256784. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant––An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Giardine, B.; Riemer, C.; Hardison, R.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2020. 2020. Available online: https://ambermd.org/doc12/Amber20.pdf (accessed on 25 July 2021).
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.B.R.; McGee, J.T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. C-Immsim 10.1 Server 2012. Available online: http://www.cbs.dtu.dk/services/C-ImmSim-10.1/ (accessed on 2 August 2021).
- Li, W.; Jia, Y. An information theoretic approach to interacting multiple model estimation. IEEE Trans. Aerosp. Electron. Syst. 2015, 51, 1811–1825. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.B.; Dombkowski, A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- ul Qamar, M.T.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; Santos, A.; Hassan, S.S.; et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio cholerae Targeted by Piper betel Derived Compounds. PLoS ONE 2013, 8, e52773. [Google Scholar] [CrossRef]
- Sheth, H.B.; Glasier, L.M.; Ellert, N.W.; Cachia, P.; Kohn, W.; Lee, K.K.; Paranchych, W.; Hodges, R.S.; Irvin, R.T. Development of an anti-adhesive vaccine for Pseudomonas aeruginosa targeting the C-terminal region of the pilin structural protein. Biomed. Pept. Proteins Nucleic Acids Struct. Synth. Boil. Act. 1995, 1, 141–148. [Google Scholar]
- Wizemann, T.M.; Adamou, J.E.; Langermann, S. Adhesins as Targets for Vaccine Development. Emerg. Infect. Dis. 1999, 5, 395–403. [Google Scholar] [CrossRef]
- Lei, Y.; Zhao, F.; Shao, J.; Li, Y.; Li, S.; Chang, H.; Zhang, Y. Application of built-in adjuvants for epitope-based vaccines. PeerJ 2019, 6, e6185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, M.T.U.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.U.; Bari, A.; Adeel, M.M.; Maryam, A.; Ashfaq, U.A.; Du, X.; Muneer, I.; Ahmad, H.I.; Wang, J. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018, 16, 298. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J. Mol. Graph. Model. 2017, 77, 72–85. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Comparative subtractive proteomics based ranking for antibiotic targets against the dirtiest superbug: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 82, 74–92. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O. V Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Transmembrane Helices | Physiochemical Properties | NCBI Blast(Human) | Antigenisity | Adhesion | Allergenisity | NCBI Blast(L. Rhamnosus) | NCBI Blast(L. Casei) | NCBI Blast(L. L. Johnsonii) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TMHMM | No of Residues | MW | Theoretical PI | Negatively Charged Residues | Positively Charged Residues | Gravy | Aliphatic Index | Instability Index | Stability | Coverage | Id | E-value | Vexijen/0.7 | Vaxign | - | Coverage | Id | E-Value | Coverage | Id | E-Value | Covarge | Id | E-Value | |
| >core/178/1/Org1_Gene2817 | 0 | 871 | 94,813.23 | 6.36 | 76 | 71 | −0.385 | 74.71 | 27.91 | Stability | no similarity | no similarity | no similarity | 0.6441 | - | - | - | - | - | - | - | - | - | - | - |
| >core/538/1/Org1_Gene4990 | 0 | 618 | 68,660.6 | 5.2 | 71 | 57 | −0.58 | 69.72 | 26.43 | Stability | no similarity | no similarity | no similarity | 0.6041 | - | - | - | - | - | - | - | - | - | - | - |
| >core/644/1/Org1_Gene1920 | 0 | 581 | 62,110.64 | 5.85 | 66 | 58 | −0.22 | 81.79 | 27.91 | Stability | 88% | 33.27% | 1.00E-80 | - | - | - | - | - | - | - | - | - | - | - | - |
| >core/870/1/Org1_Gene3696 | 0 | 528 | 55,681.97 | 5.9 | 51 | 39 | −0.071 | 91.27 | 42.67 | unstable | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| >core/5642/1/Org1_Gene690 | 0 | 206 | 22,970.88 | 6.23 | 25 | 22 | −0.386 | 77.38 | 31.68 | Stability | 100% | 43.60% | 5.00E-56 | - | - | - | - | - | - | - | - | - | - | - | - |
| >core/5897/1/Org1_Gene256 | 0 | 193 | 21,147.69 | 5.76 | 19 | 14 | −0.141 | 78.08 | 22.96 | Stability | 100% | 43.14% | 2.00E-47 | - | - | - | - | - | - | - | - | - | - | - | - |
| >core/6413/1/Org1_Gene5320 | 0 | 161 | 16,537.39 | 5.15 | 10 | 8 | 0.096 | 89.69 | 32.37 | Stability | no similarity | no similarity | no similarity | 0.9532 | 0.965 | ALLERGEN | - | - | - | - | - | - | - | - | - |
| >core/178/3/Org3_Gene3169 | 0 | 878 | 94,683.95 | 6.04 | 75 | 68 | −0.366 | 75.02 | 24.93 | Stability | no similarity | no similarity | no similarity | 0.6563 | - | - | - | - | - | - | - | - | - | - | - |
| >core/276/3/Org3_Gene2971 | 0 | 789 | 86,600.87 | 5.41 | 81 | 66 | −0.583 | 60.04 | 31.03 | Stability | no similarity | no similarity | no similarity | 0.6852 | - | - | - | - | - | - | - | - | - | - | - |
| >core/325/3/Org3_Gene4813 | 0 | 753 | 82,490.82 | 5.72 | 75 | 62 | −0.573 | 66.99 | 29.48 | Stability | no similarity | no similarity | no similarity | 0.8125 | 0.856 | NONALLERGEN | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity |
| >core/538/3/Org3_Gene4136 | 0 | 618 | 68,572.26 | 5.07 | 69 | 51 | −0.586 | 69.72 | 25.21 | Stability | no similarity | no similarity | no similarity | 0.625 | - | - | - | - | - | - | - | - | - | - | - |
| >core/870/3/Org3_Gene994 | 0 | 527 | 55,704 | 5.75 | 53 | 40 | −0.089 | 89.39 | 37.5 | Stability | 95% | 27.03% | 2.00E-21 | 0.4053 | - | - | - | - | - | - | - | - | - | - | - |
| >core/538/4/Org4_Gene3945 | 0 | 622 | 69,756.5 | 5.22 | 71 | 56 | −0.668 | 64.55 | 25.01 | Stability | no similarity | no similarity | no similarity | 0.5795 | - | - | - | - | - | - | - | - | - | - | - |
| >core/3258/4/Org4_Gene4637 | 0 | 300 | 31,253.99 | 9.01 | 10 | 15 | 0.064 | 85.13 | 31.98 | Stability | no similarity | no similarity | no similarity | 0.7627 | 0.908 | ALLERGEN | - | - | - | - | - | - | - | - | - |
| >core/6413/4/Org4_Gene2075 | 0 | 167 | 17,112.08 | 5.03 | 9 | 7 | 0.095 | 84.73 | 23.68 | Stability | no similarity | no similarity | no similarity | 0.8816 | 0.959 | ALLERGEN | - | - | - | - | - | - | - | - | - |
| >core/591/6/Org6_Gene4047 | 0 | 597 | 61,743.78 | 7.64 | 37 | 38 | −0.123 | 85.28 | 15.68 | Stability | no similarity | no similarity | no similarity | 0.6161 | - | ALLERGEN | |||||||||
| >core/325/7/Org7_Gene2597 | 0 | 742 | 82,442.17 | 5.42 | 86 | 73 | −0.68 | 64.78 | 33.5 | Stability | no similarity | no similarity | no similarity | 0.8217 | 0.807 | NONALLERGEN | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity | no similarity |
| >core/538/7/Org7_Gene4577 | 0 | 615 | 68,043.77 | 5.17 | 66 | 51 | −0.481 | 70.99 | 26.7 | Stability | no similarity | no similarity | no similarity | 0.5512 | - | ALLERGEN | - | - | - | - | - | - | - | - | - |
| >core/870/7/Org7_Gene4277 | 0 | 527 | 56,010.35 | 6.05 | 49 | 40 | −0.117 | 89.73 | 41.54 | unstable | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Proteins | B-Cell Epitopes | T-Cell Epitopes | Percentile Rank | MHC II | Percentile Rank | Common Peptides | Antigenicity/0.5 | Allergenicity | Solubility | MHC Pred | Toxicity | Virulent | IFN Gamma | Final | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| >core/325/3/Org3_Gene4813 (TonB-dependent siderophore receptor) | YGRQKPKRFNYKGESVSGSELNEV | RQKPKRFNY | 1.3 | RQKPKRFNYKGESVS | 54 | RQKPKRFNY | 0.9784 | Antigen | nonallergen | Good water solubility | 2.37 | nontoxin | Virulent | positive | Selected |
| YSRQGNLYAGDTQNTNTSTLVKSMYGKETNRLY | NTNTSTLVK | 2.3 | QNTNTSTLVKSMYGK | 55 | NTNTSTLVK | 0.7856 | Antigen | nonallergen | Good water solubility | 55.98 | nontoxin | Virulent | positive | Selected | |
| SKTQADAQDINSGHEAARTGSYAGSYPAGREGVVNKDIHG | QADAQDINS | 7.5 | QADAQDINSGHEAAR | 85 | QADAQDINS | 1.4865 | Antigen | nonallergen | Good water solubility | 4.83 | nontoxin | Virulent | positive | Selected | |
| >core/325/7/Org7_Gene2597 (siderophore enterobactin receptor FepA) | SRQGNLYAGDTQNTNSNDLVKENYGKETNRLYR | SNDLVKENY | 0.24 | SNDLVKENYGKETNRLYR | 59 | SNDLVKENY | 0.8435 | Antigen | nonallergen | Good water solubility | 46.99 | nontoxin | Virulent | positive | Selected |
| QTNPNYILYSKGQGCYASKSGCYLQGNDDLKAE | YLQGNDDLK | 3.4 | GCYLQGNDDLKA | 41 | YLQGNDDLK | 1.4275 | Antigen | nonallergen | Good water solubility | 21.73 | nontoxin | Virulent | positive | Selected | |
| LDKTQADAWDINQGHQSERTGIYADTLPAGREGVE | QSERTGIYA | 1.7 | QSERTGIYADTL | 55 | QSERTGIYA | 0.5139 | Antigen | nonallergen | Good water solubility | 33.19 | nontoxin | Virulent | positive | Selected | |
| QADAWDINQ | 4.5 | QADAWDINQGHQS | 71 | QADAWDINQ | 0.8036 | Antigen | nonallergen | Good water solubility | 10.09 | nontoxin | Virulent | positive | Selected | ||
| Model | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored | GALAXY Energy |
|---|---|---|---|---|---|---|
| Initial | 0 | 3.91 | 109.2 | 9.7 | 84.5 | 24,937.41 |
| MODEL 1 | 0.89 | 1.48 | 1.9 | 0.6 | 90.5 | −3648.56 |
| MODEL 2 | 1.29 | 1.56 | 3 | 0 | 92.3 | −3646.33 |
| MODEL 3 | 0.88 | 1.50 | 2.4 | 0 | 92.3 | −3638.63 |
| MODEL 4 | 0.88 | 1.35 | 1.4 | 0 | 91.8 | −3624.25 |
| MODEL 5 | 0.95 | 1.47 | 1.9 | 0 | 90.9 | −3623.95 |
| MODEL 6 | 0.85 | 1.56 | 2.4 | 0 | 90.5 | −3618.86 |
| MODEL 7 | 1.65 | 1.57 | 3 | 0 | 91.8 | −3617.76 |
| MODEL 8 | 2.75 | 1.62 | 3.2 | 0 | 91.4 | −3617.33 |
| MODEL 9 | 1.45 | 1.51 | 2.7 | 0 | 92.7 | −3616.55 |
| MODEL 10 | 1.05 | 1.66 | 3.5 | 0.6 | 90.9 | −3616.14 |
| TLR4 | ||||||
|---|---|---|---|---|---|---|
| Rank | Solution Number | Global Energy | Attractive van der Waals Energy | Repulsive van der Waals Energy | Atomic Contact Energy | Hydrogen Bonds Energy |
| 1 | 4 | −16.01 | −31.00 | 8.99 | 10.72 | −4.13 |
| 2 | 2 | −8.24 | −30.86 | 13.13 | 15.55 | −4.23 |
| 3 | 1 | −3.81 | −33.73 | 10.52 | 21.45 | −6.01 |
| 4 | 10 | 4.69 | −45.43 | 21.19 | 20.21 | −1.33 |
| 5 | 5 | 8.46 | −24.26 | 14.75 | 19.26 | −1.50 |
| 6 | 9 | 10.47 | −0.37 | 0.00 | 1.03 | 0.00 |
| 7 | 6 | 17.68 | −18.48 | 22.97 | 8.59 | −1.86 |
| 8 | 7 | 23.80 | −20.71 | 10.74 | 11.10 | −0.81 |
| 9 | 3 | 31.16 | −12.53 | 25.41 | 11.03 | −0.30 |
| 10 | 8 | 44.88 | −21.53 | 7.66 | 10.71 | −2.45 |
| MHC-I | ||||||
| Rank | Solution Number | Global Energy | Attractive van der Waals Energy | Repulsive van der Waals Energy | Atomic Contact Energy | Hydrogen Bonds Energy |
| 1 | 7 | −2.22 | −2.06 | 0.04 | 0.98 | −0.50 |
| 2 | 9 | 12.40 | −0.55 | 0.00 | 0.34 | 0.00 |
| 3 | 8 | 13.34 | −31.79 | 53.87 | 6.76 | −5.68 |
| 4 | 4 | 48.19 | −31.31 | 94.84 | 6.68 | −8.33 |
| 5 | 2 | 603.03 | −55.26 | 811.32 | 5.43 | −5.36 |
| 6 | 10 | 654.52 | −25.48 | 850.38 | 7.08 | −5.82 |
| 7 | 5 | 697.54 | −49.00 | 968.22 | −2.34 | −3.14 |
| 8 | 3 | 1338.16 | −36.09 | 1705.36 | 9.00 | −6.47 |
| 9 | 1 | 1394.24 | −29.34 | 1801.68 | −2.37 | −1.68 |
| 10 | 6 | 4166.32 | −67.09 | 5348.85 | −7.31 | −7.27 |
| MHC−II | ||||||
| Rank | Solution Number | Global Energy | Attractive van der Waals Energy | Repulsive van der Waals Energy | Atomic Contact Energy | Hydrogen Bonds Energy |
| 1 | 7 | −39.32 | −35.52 | 14.84 | 6.51 | −2.57 |
| 2 | 9 | −32.36 | −28.35 | 9.77 | −1.69 | −0.95 |
| 3 | 4 | −25.80 | −29.69 | 12.42 | 15.73 | −4.94 |
| 4 | 2 | −18.97 | −41.15 | 34.17 | 3.31 | −5.99 |
| 5 | 1 | −9.05 | −7.99 | 1.86 | 1.04 | −0.31 |
| 6 | 8 | 3.14 | −13.88 | 1.90 | 6.13 | −0.67 |
| 7 | 10 | 7.17 | −22.42 | 45.49 | −0.73 | −1.11 |
| 8 | 3 | 13.32 | −0.85 | 0.00 | −0.73 | 0.00 |
| 9 | 5 | 13.70 | −2.59 | 1.00 | 2.27 | −0.41 |
| 10 | 6 | 1319.04 | −56.48 | 1760.85 | 6.51 | −6.93 |
| MMGBSA | MMPBSA | ||
|---|---|---|---|
| TLR4 Vaccine Complex | |||
| Energy Component | Average | Energy Component | Average |
| VDWALLS | −324.47 | VDWALLS | −324.47 |
| EEL | −172.48 | EEL | −172.48 |
| EGB | −198.69 | EPB | −159.57 |
| ESURF | −18.64 | ENPOLAR | −22.00 |
| Delta G gas | −496.95 | Delta G gas | −496.95 |
| Delta G solve | −217.33 | Delta G solve | −181.57 |
| Total | −714.28 | Total | −678.52 |
| MHC-I Vaccine Complex | |||
| Energy Component | Average | Energy Component | Average |
| VDWALLS | −311.42 | VDWALLS | −311.42 |
| EEL | −172.58 | EEL | −172.58 |
| EGB | −105.66 | EPB | −147.83 |
| ESURF | −21.03 | ENPOLAR | −17.53 |
| Delta G gas | −484 | Delta G gas | −484 |
| Delta G solve | −126.69 | Delta G solve | −165.36 |
| Total | −610.69 | Total | −649.36 |
| MHC-II Vaccine Complex | |||
| Energy Component | Average | Energy Component | Average |
| VDWALLS | −80.35 | VDWALLS | −80.35 |
| EEL | 90.16 | EEL | 90.16 |
| EGB | −67.33 | EPB | −73.09 |
| ESURF | −15.77 | ENPOLAR | −11.54 |
| Delta G gas | 9.81 | Delta G gas | 9.81 |
| Delta G solve | −83.1 | Delta G solve | −84.63 |
| Total | −73.29 | Total | −74.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allemailem, K.S. A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques. Vaccines 2021, 9, 1087. https://doi.org/10.3390/vaccines9101087
Allemailem KS. A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques. Vaccines. 2021; 9(10):1087. https://doi.org/10.3390/vaccines9101087
Chicago/Turabian StyleAllemailem, Khaled S. 2021. "A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques" Vaccines 9, no. 10: 1087. https://doi.org/10.3390/vaccines9101087