
A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Line, Culture Conditions, and Transient Gene Expression
2.2. Spectrofluorometry
2.3. Flow Virometry
2.4. Total Protein, Host Cell Protein (HCP), and Double-Stranded DNA (dsDNA) Quantification
2.5. Dynamic Light Scattering (DLS)
2.6. Nanoparticle Tracking Analysis (NTA)
2.7. Cryogenic Transmission Electron Microscopy (Cryo-TEM)
2.8. Sodium Dodecyl Sulfate-Polyacrylamide Gel (SDS-PAGE) Electrophoresis and Western Blot Analysis
2.9. Thermostability Studies of Gag-eGFP VLPs
2.10. Preliminary Serial Filtration Experiments
2.11. Preparative Clarification Experiments
2.12. Ion-Exchange Chromatography (IEX)
2.13. SEC Chromatography
2.14. Lyophilization
3. Results
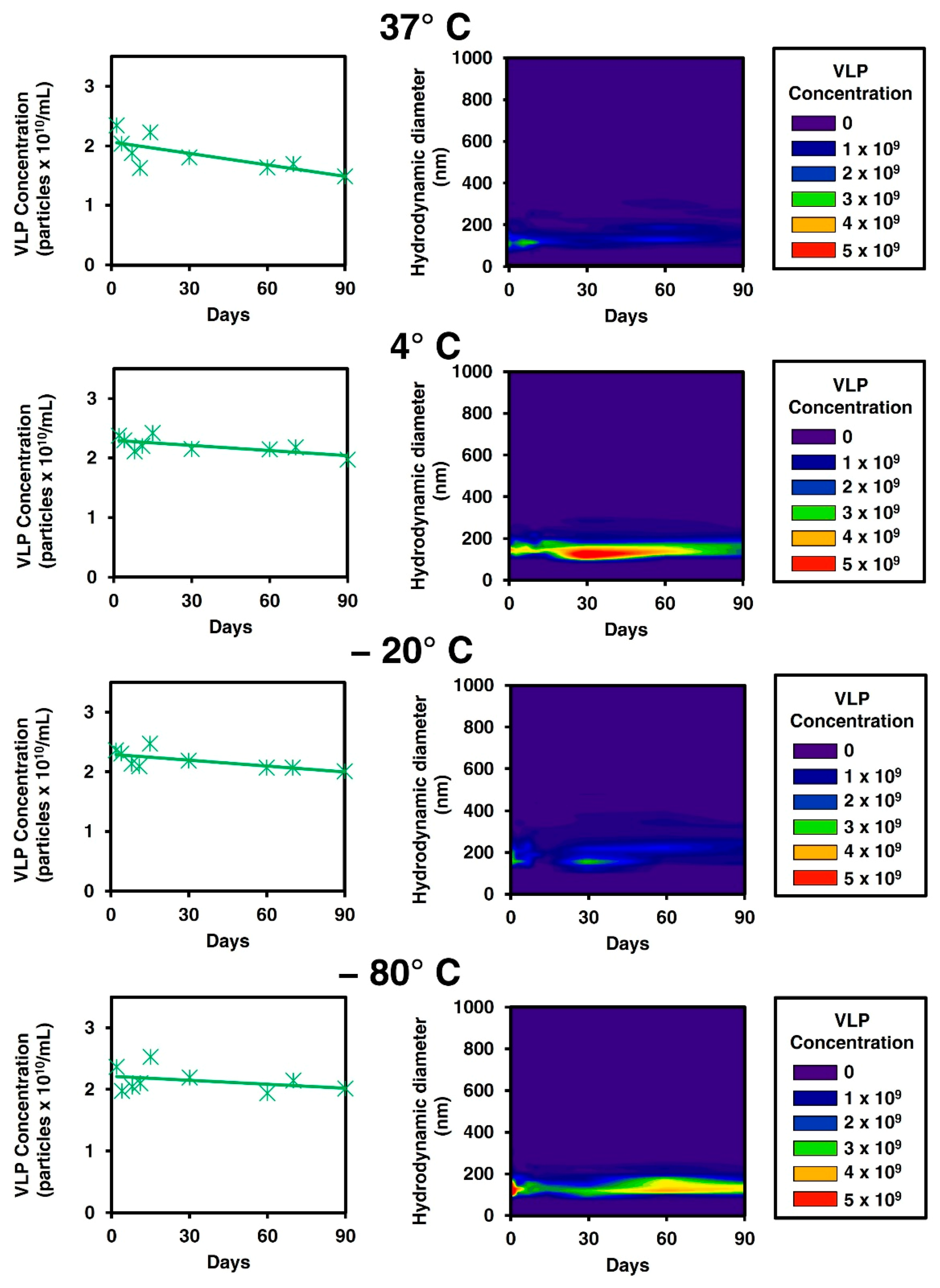
3.1. Stability Studies on Gag VLPs
3.2. Development of Four Steps Downstream Process of Gag VLPs
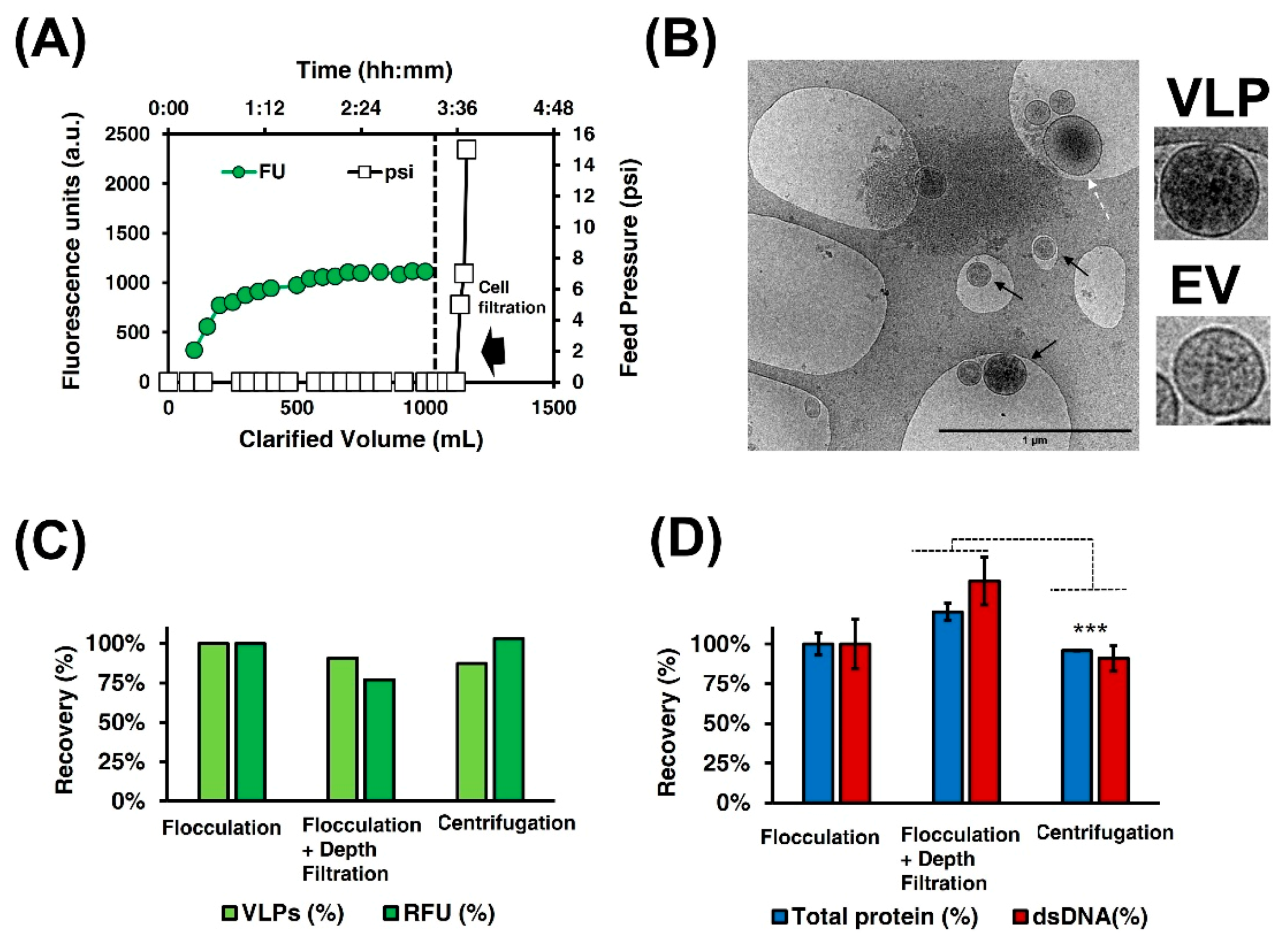
3.2.1. Clarification
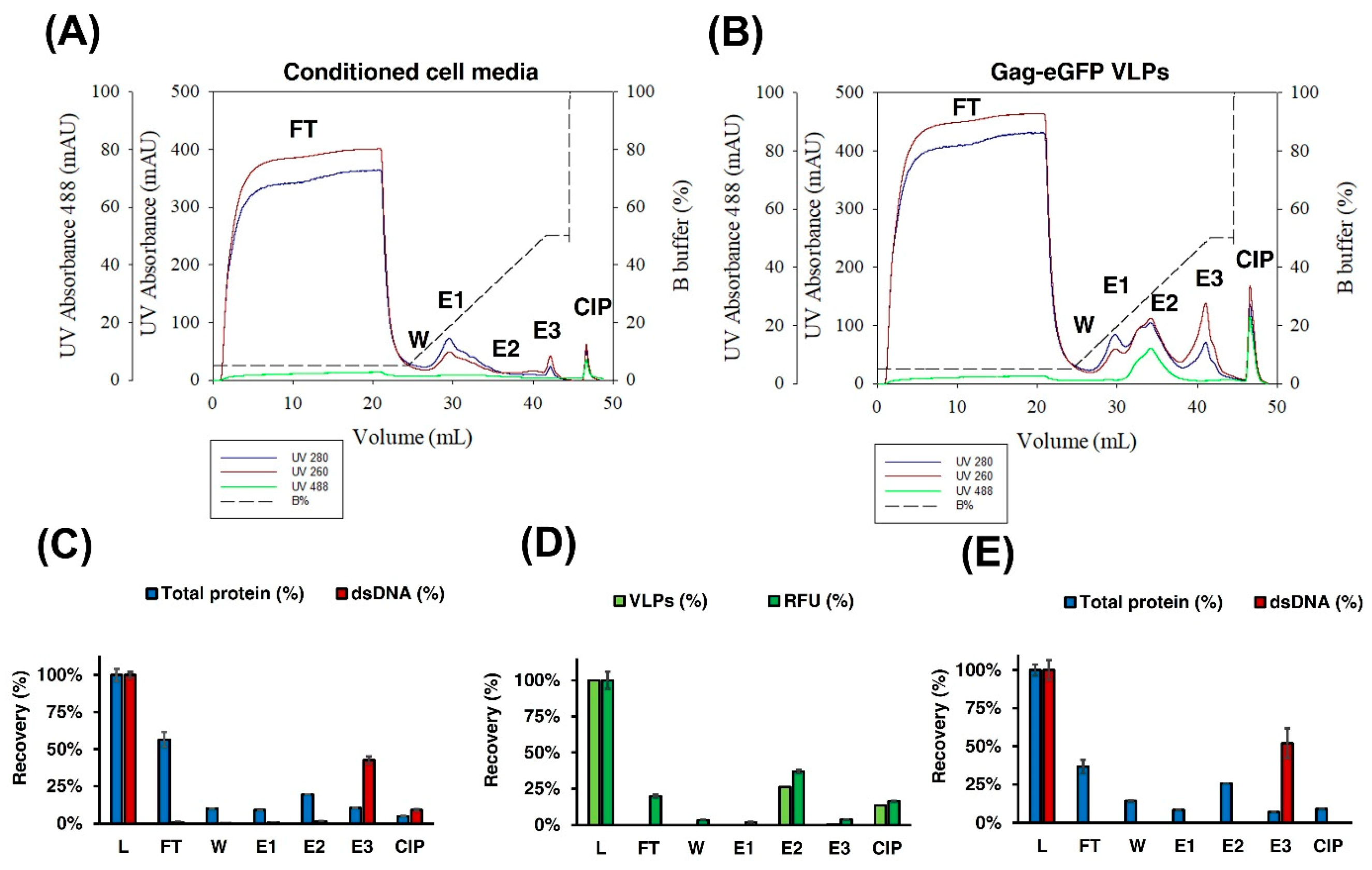
3.2.2. IEX Chromatography
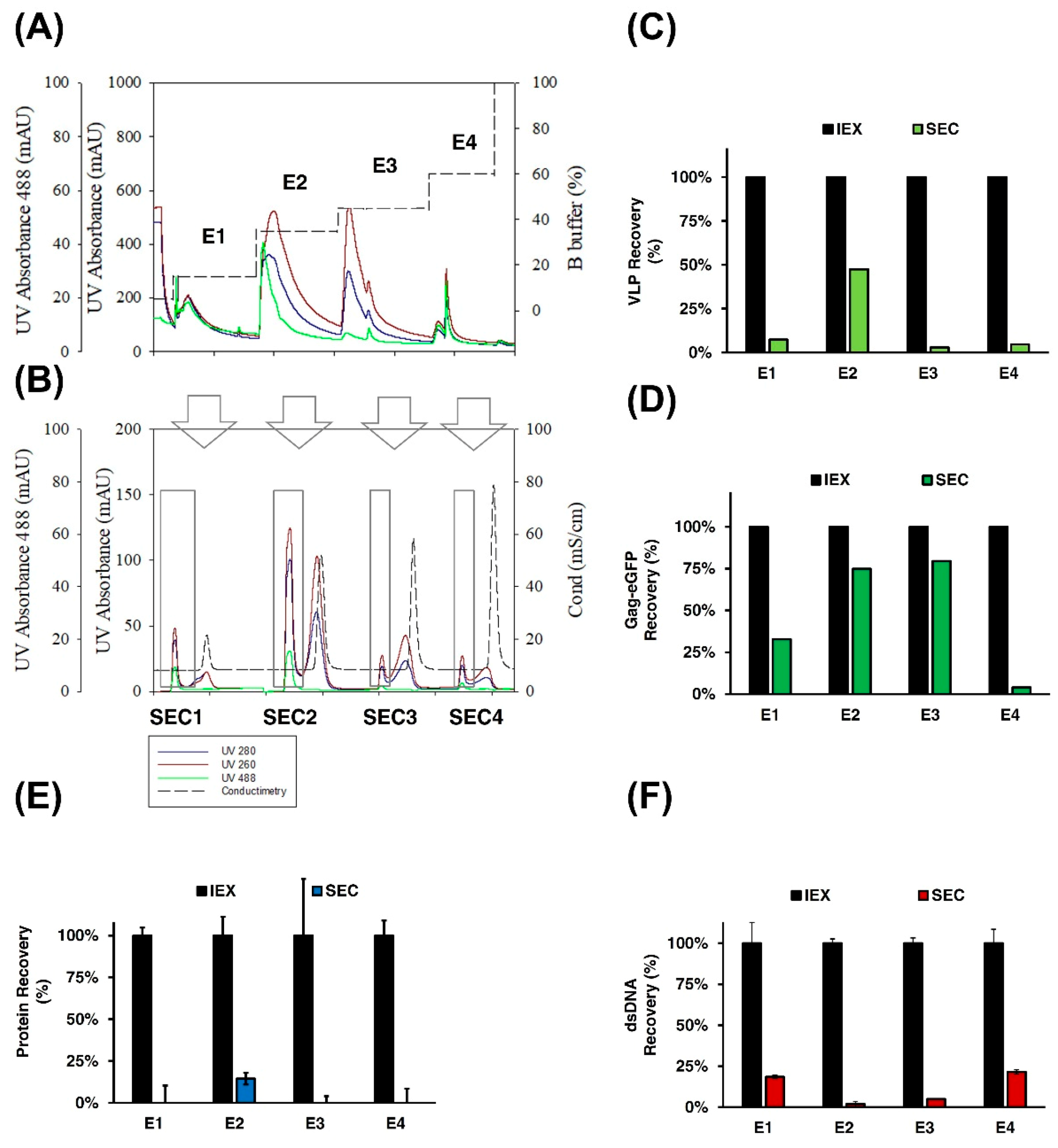
3.2.3. SEC Chromatography
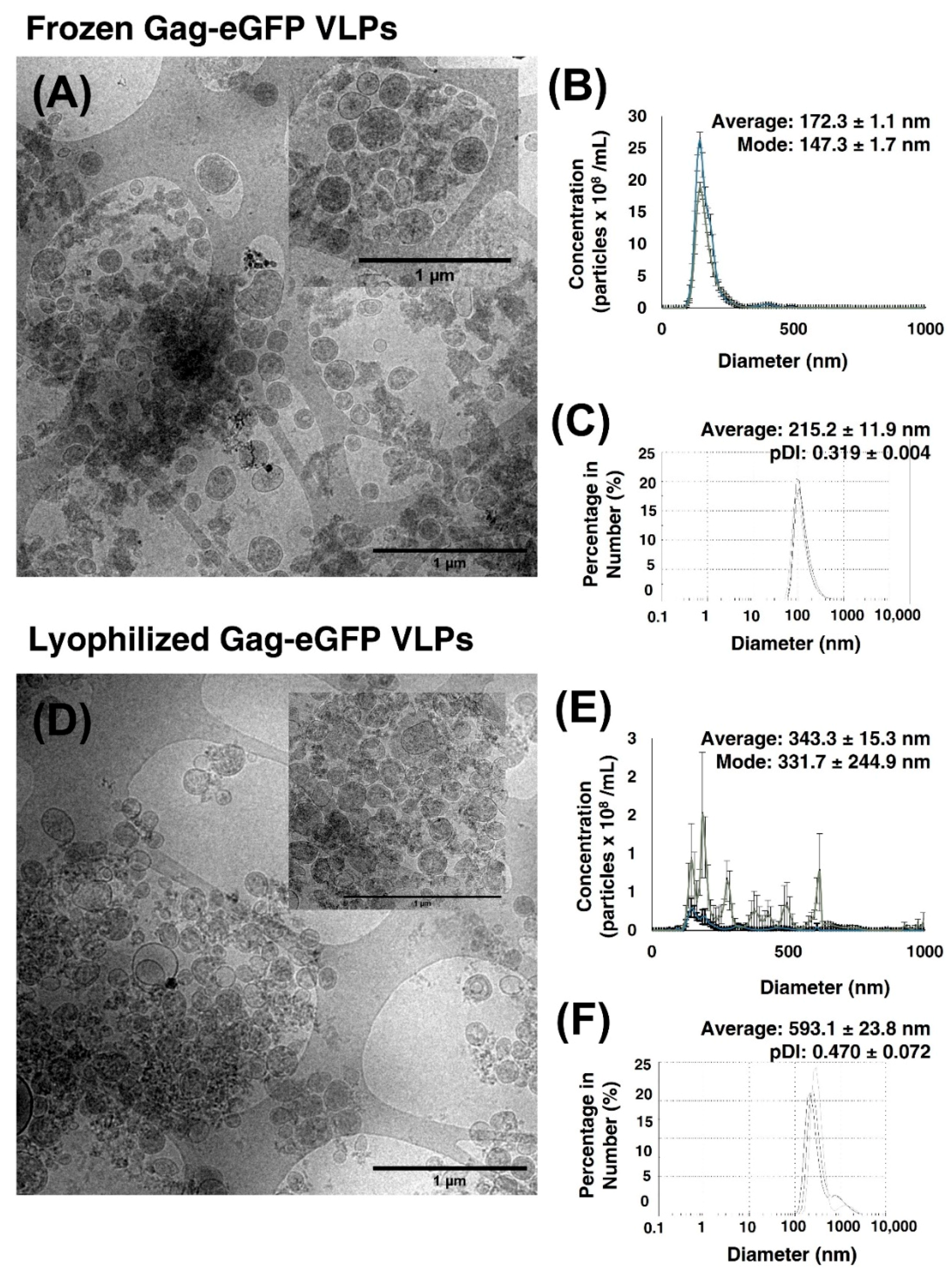
3.2.4. Lyophilization of Gag-eGFP VLPs
4. Discussion
4.1. Purification Train and Unit Capacity
4.2. VLP Quality and Purification Yield
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charlton Hume, H.K.; Vidigal, J.; Carrondo, M.J.T.; Middelberg, A.P.J.; Roldão, A.; Lua, L.H.L. Synthetic biology for bioengineering virus-like particle vaccines. Biotechnol. Bioeng. 2019, 116, 919–935. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formulation for clinical translation. Expert Rev. Vaccines 2018, 17, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.D.; Zak, A.; Khera, E.; Wen, F. Engineering Virus-like Particles for Antigen and Drug Delivery. Curr. Protein Pept. Sci. 2017, 19, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Cervera, L.; Gòdia, F.; Tarrés-Freixas, F.; Aguilar-Gurrieri, C.; Carrillo, J.; Blanco, J.; Gutiérrez-Granados, S. Production of HIV-1-based virus-like particles for vaccination: Achievements and limits. Appl. Microbiol. Biotechnol. 2019, 103, 7367–7384. [Google Scholar] [CrossRef] [PubMed]
- Göttlinger, H.G. HIV-1 Gag: A Molecular Machine Driving Viral Particle Assembly and Release; Kuiken, C.K., Foley, B., Leitner, T., Apetrei, C., Hahn, B., Mizrachi, I., Mullins, J., Rambaut, A., StevenWolinsky, S., Eds.; Los Alamos National Laboratory, Theoretical Biology and Biophysics: Los Alamos, NM, USA, 2001; ISBN 0269-9370. [Google Scholar]
- Besnard, L.; Fabre, V.; Fettig, M.; Gousseinov, E.; Kawakami, Y.; Laroudie, N.; Scanlan, C.; Pattnaik, P. Clarification of vaccines: An overview of filter based technology trends and best practices. Biotechnol. Adv. 2016, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Effio, C.L.; Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015, 10, 715–727. [Google Scholar] [CrossRef]
- Moleirinho, M.G.; Silva, R.J.S.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Current challenges in biotherapeutic particles manufacturing. Expert Opin. Biol. Ther. 2020, 20, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Pato, T.P.; Souza, M.C.O.; Silva, A.N.M.R.; Pereira, R.C.; Silva, M.V.; Caride, E.; Gaspar, L.P.; Freire, M.S.; Castilho, L.R. Development of a membrane adsorber based capture step for the purification of yellow fever virus. Vaccine 2014, 32, 2789–2793. [Google Scholar] [CrossRef] [Green Version]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Aguilar, P.P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nöbauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Pereira Aguilar, P.; Schneider, T.A.; Wetter, V.; Maresch, D.; Ling, W.L.; Tover, A.; Steppert, P.; Jungbauer, A. Polymer-grafted chromatography media for the purification of enveloped virus-like particles, exemplified with HIV-1 gag VLP. Vaccine 2019, 37, 7070–7080. [Google Scholar] [CrossRef]
- Aguilar, P.P.; Reiter, K.; Wetter, V.; Steppert, P.; Maresch, D.; Ling, W.L.; Satzer, P.; Jungbauer, A. Capture and purification of Human Immunodeficiency Virus-1 virus-like particles: Convective media vs. porous beads. J. Chromatogr. A 2020, 1627, 461378. [Google Scholar] [CrossRef] [PubMed]
- Reiter, K.; Aguilar, P.P.; Wetter, V.; Steppert, P.; Tover, A.; Jungbauer, A. Separation of virus-like particles and extracellular vesicles by flow-through and heparin affinity chromatography. J. Chromatogr. A 2019, 1588, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.B.; Silva, R.J.S.; Moleirinho, M.G.; Cunha, B.; Moreira, A.S.; Xenopoulos, A.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Membrane-Based Approach for the Downstream Processing of Influenza Virus-Like Particles. Biotechnol. J. 2019, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Venereo-Sanchez, A.; Gilbert, R.; Simoneau, M.; Caron, A.; Chahal, P.; Chen, W.; Ansorge, S.; Li, X.; Henry, O.; Kamen, A. Hemagglutinin and neuraminidase containing virus-like particles produced in HEK-293 suspension culture: An effective influenza vaccine candidate. Vaccine 2016, 34, 3371–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Kramberger, P.; Tover, A.; Berger, E.; Nöbauer, K.; Razzazi-Fazeli, E.; Jungbauer, A. Separation of HIV-1 gag virus-like particles from vesicular particles impurities by hydroxyl-functionalized monoliths. J. Sep. Sci. 2017, 40, 979–990. [Google Scholar] [CrossRef]
- Gutiérrez-Granados, S.; Cervera, L.; Gòdia, F.; Carrillo, J.; Segura, M.M.; Gutierrez-Granados, S.; Cervera, L.; Godia, F.; Carrillo, J.; Segura, M.M.; et al. Development and validation of a quantitation assay for fluorescently tagged HIV-1 virus-like particles. J. Virol. Methods 2013, 193, 85–95. [Google Scholar] [CrossRef]
- González-Domínguez, I.; Puente-Massaguer, E.; Cervera, L.; Gòdia, F. Quality Assessment of Virus-Like Particles at Single Particle Level: A Comparative Study. Viruses 2020, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Cervera, L.; González-Domínguez, I.; Segura, M.M.; Gòdia, F. Intracellular characterization of Gag VLP production by transient transfection of HEK 293 cells. Biotechnol. Bioeng. 2017, 114, 2507–2517. [Google Scholar] [CrossRef]
- Kumru, O.S.; Joshi, S.B.; Smith, D.E.; Middaugh, C.R.; Prusik, T.; Volkin, D.B. Vaccine instability in the cold chain: Mechanisms, analysis and formulation strategies. Biologicals 2014, 42, 237–259. [Google Scholar] [CrossRef] [Green Version]
- Lynch, A.; Meyers, A.E.; Williamson, A.-L.; Rybicki, E.P. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol. J. 2012, 9, 210. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.J.J.; Daoussi, R.; Vervaet, C.; Remon, J.P.; De Beer, T.R.M. Freeze-drying of live virus vaccines: A review. Vaccine 2015, 33, 5507–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervera, L.; Gutiérrez-Granados, S.; Martínez, M.; Blanco, J.; Gòdia, F.; Segura, M.M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. J. Biotechnol. 2013, 166, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, B.; Musier-Forsyth, K.; Mansky, L.M.; Mueller, J.D. Fluorescence fluctuation spectroscopy on viral-like particles reveals variable Gag stoichiometry. Biophys. J. 2009, 96, 1961–1969. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, I.; Grimaldi, N.; Cervera, L.; Ventosa, N.; Gòdia, F. Impact of physicochemical properties of DNA/PEI complexes on transient transfection of mammalian cells. New Biotechnol. 2019, 49, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.R.H.; Cervera, L.; Ahmed, I.; Kondratov, O.; Zolotukhin, S.; Schrag, J.; Chahal, P.S.; Kamen, A.A. Achieving High-Yield Production of Functional AAV5 Gene Delivery Vectors via Fedbatch in an Insect Cell-One Baculovirus System. Mol. Ther. Methods Clin. Dev. 2019, 13, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Pereira Aguilar, P.; González-Domínguez, I.; Schneider, T.A.T.A.; Gòdia, F.; Cervera, L.; Jungbauer, A.; González-Domínguez, I.; Schneider, T.A.T.A.; Gòdia, F.; Cervera, L.; et al. At-line multi-angle light scattering detector for faster process development in enveloped virus-like particle purification. J. Sep. Sci. 2019, 42, 2640–2648. [Google Scholar]
- Cervera, L.; Fuenmayor, J.; González-Domínguez, I.; Gutiérrez-Granados, S.; Segura, M.M.; Gòdia, F. Selection and optimization of transfection enhancer additives for increased virus-like particle production in HEK293 suspension cell cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9935–9949. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Silva, R.J.S.; Moreira, A.S.; Cunha, B.; Clemente, J.J.; Alves, P.M.; Carrondo, M.J.T.; Xenopoulos, A.; Peixoto, C. Efficient filtration strategies for the clarification of influenza virus-like particles derived from insect cells. Sep. Purif. Technol. 2019, 218, 81–88. [Google Scholar] [CrossRef]
- Malvern Instruments Limited. Determining Fluorescence Limit of Detection with Nanoparticle Tracking Analysis (NTA); Malvern Instruments Limited: Malvern, UK, 2015. [Google Scholar]
- González-Domínguez, I.; Puente-Massaguer, E.; Cervera, L.; Gòdia, F. Quantification of the HIV-1 virus-like particle production process by super-resolution imaging: From VLP budding to nanoparticle analysis. Biotechnol. Bioeng. 2020, 117, 1929–1945. [Google Scholar] [CrossRef]
- Anderson, E.C.; Petersen, D.F.; Tobey, R.A. Density Invariance of Cultured Chinese Hamster Cells with Stage of the Mitotic Cycle. Biophys. J. 1970, 10, 630–645. [Google Scholar] [CrossRef] [Green Version]
- Westoby, M.; Chrostowski, J.; De Vilmorin, P.; Smelko, J.P.; Romero, J.K.; Carolina, N. Effects of Solution Environment on Mammalian Cell Fermentation Broth Properties: Enhanced Impurity Removal and Clarification Performance. Biotechnol. Bioeng. 2011, 108, 50–58. [Google Scholar] [CrossRef]
- Nestola, P.; Villain, L.; Peixoto, C.; Martins, D.L.; Alves, P.M.; Carrondo, M.J.T.; Mota, J.P.B. Impact of grafting on the design of new membrane adsorbers for adenovirus purification. J. Biotechnol. 2014, 181, 1–11. [Google Scholar] [CrossRef]
- McNally, D.J.J.; Darling, D.; Farzaneh, F.; Levison, P.R.R.; Slater, N.K.H.K.H. Optimised concentration and purification of retroviruses using membrane chromatography. J. Chromatogr. A 2014, 1340, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutner, R.H.; Puthli, S.; Marino, M.P.; Reiser, J. Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol. 2009, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestola, P.; Peixoto, C.; Silva, R.R.J.S.; Alves, P.M.; Mota, J.P.B.; Carrondo, M.J.T. Improved virus purification processes for vaccines and gene therapy. Biotechnol. Bioeng. 2015, 112, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Venereo-Sanchez, A.; Simoneau, M.; Lanthier, S.; Chahal, P.; Bourget, L.; Ansorge, S.; Gilbert, R.; Henry, O.; Kamen, A. Process intensification for high yield production of influenza H1N1 Gag virus-like particles using an inducible HEK-293 stable cell line. Vaccine 2017, 35, 4220–4228. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, I.; Stacey, G.; Petricciani, J. WHO Study Group on cell substrates for production of biologicals, Geneva, Switzerland, 11–12 June 2007. Biologicals 2008, 36, 203–211. [Google Scholar] [CrossRef]
- Eaton, L.C. Host cell contaminant protein assay development for recombinant biopharmaceuticals. J. Chromatogr. A 1995, 705, 105–114. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Volume (mL) | Total Protein | Host Cell Protein | dsDNA | Gag-eGFP Protein | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (µg/mL) | Recovery (%) | Concentration (µg/mL) | Recovery (%) | Concentration (ng/mL) | Recovery (%) | Concentration (µg/mL) | Recovery (%) | Gag-eGFP/Total Protein (%) | ||
| CL | 235 | 707.8 | 100 | 1.5 | 100 | 657.0 | 100 | 9.5 | 100 | 1 |
| IEX | 6 | 540.9 | 2 | 2.5 | 4 | 1610.7 | 6 | 99.1 | 27 | 18 |
| SEC | 10 | 38.5 | 0 | 3.1 | 9 | 31.8 | 0 | 37.8 | 17 | 98 |
| Sample | Volume (mL) | NTA (Particles × 109/mL) | VLP Recovery (%) | EV Recovery (%) | VLPs/ Total Particles (%) | |
|---|---|---|---|---|---|---|
| VLPs | EVs | |||||
| Cl | 235 | 8.2 | 23.8 | 100 | 100 | 26 |
| IEX | 6 | 117.0 | 238.0 | 37 | 25 | 36 |
| SEC | 10 | 43.3 | 34.3 | 23 | 6 | 56 |
| Process | Maximum Volume (mL) | Effective Surface Area|Volume | Load Capacity | Concentration Factor | Flux Rate | |||
|---|---|---|---|---|---|---|---|---|
| Area (cm2) | Volume (mL) | (L/m2) | (mL/mL) | LMH | mL/min | |||
| Primary Filtration | 1000 | 23 | - | 434.8 | - | 1 | 135.5 | - |
| IEX Chromatography | 413 | 4.9 | 0.86 | 842.9 | 480.2 | 50–55 | - | 1–5 |
| SEC Chromatography | 6.4 | 2.01 | 43 | - | 0.2 | 0.6 | - | 1–2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Domínguez, I.; Lorenzo, E.; Bernier, A.; Cervera, L.; Gòdia, F.; Kamen, A. A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles. Vaccines 2021, 9, 1154. https://doi.org/10.3390/vaccines9101154
González-Domínguez I, Lorenzo E, Bernier A, Cervera L, Gòdia F, Kamen A. A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles. Vaccines. 2021; 9(10):1154. https://doi.org/10.3390/vaccines9101154
Chicago/Turabian StyleGonzález-Domínguez, Irene, Elianet Lorenzo, Alice Bernier, Laura Cervera, Francesc Gòdia, and Amine Kamen. 2021. "A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles" Vaccines 9, no. 10: 1154. https://doi.org/10.3390/vaccines9101154
APA StyleGonzález-Domínguez, I., Lorenzo, E., Bernier, A., Cervera, L., Gòdia, F., & Kamen, A. (2021). A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles. Vaccines, 9(10), 1154. https://doi.org/10.3390/vaccines9101154