
Development of a Candidate Multi-Epitope Subunit Vaccine against Klebsiella aerogenes: Subtractive Proteomics and Immuno-Informatics Approach

 , , ,
, , ,  and
and
Abstract
:1. Introduction
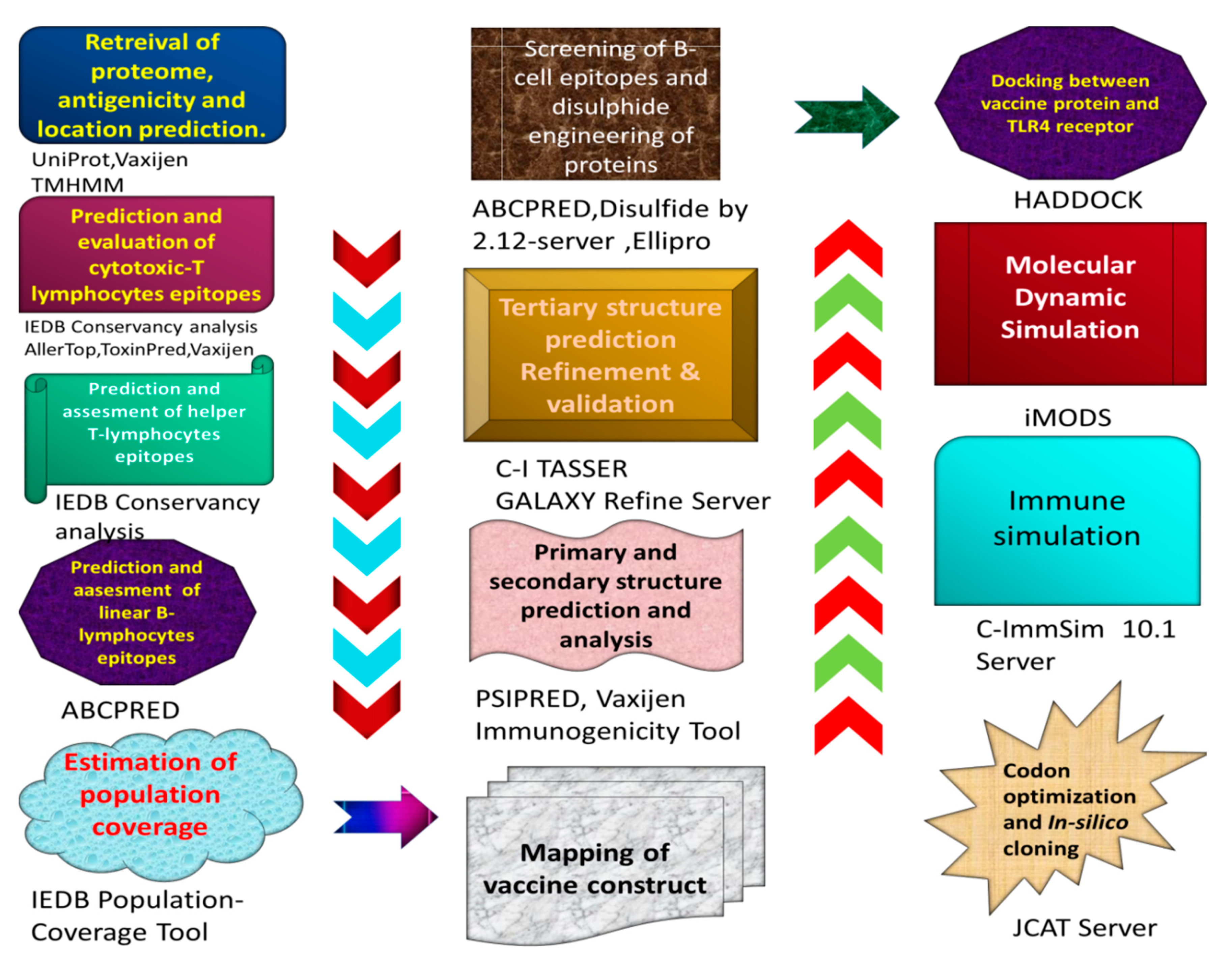
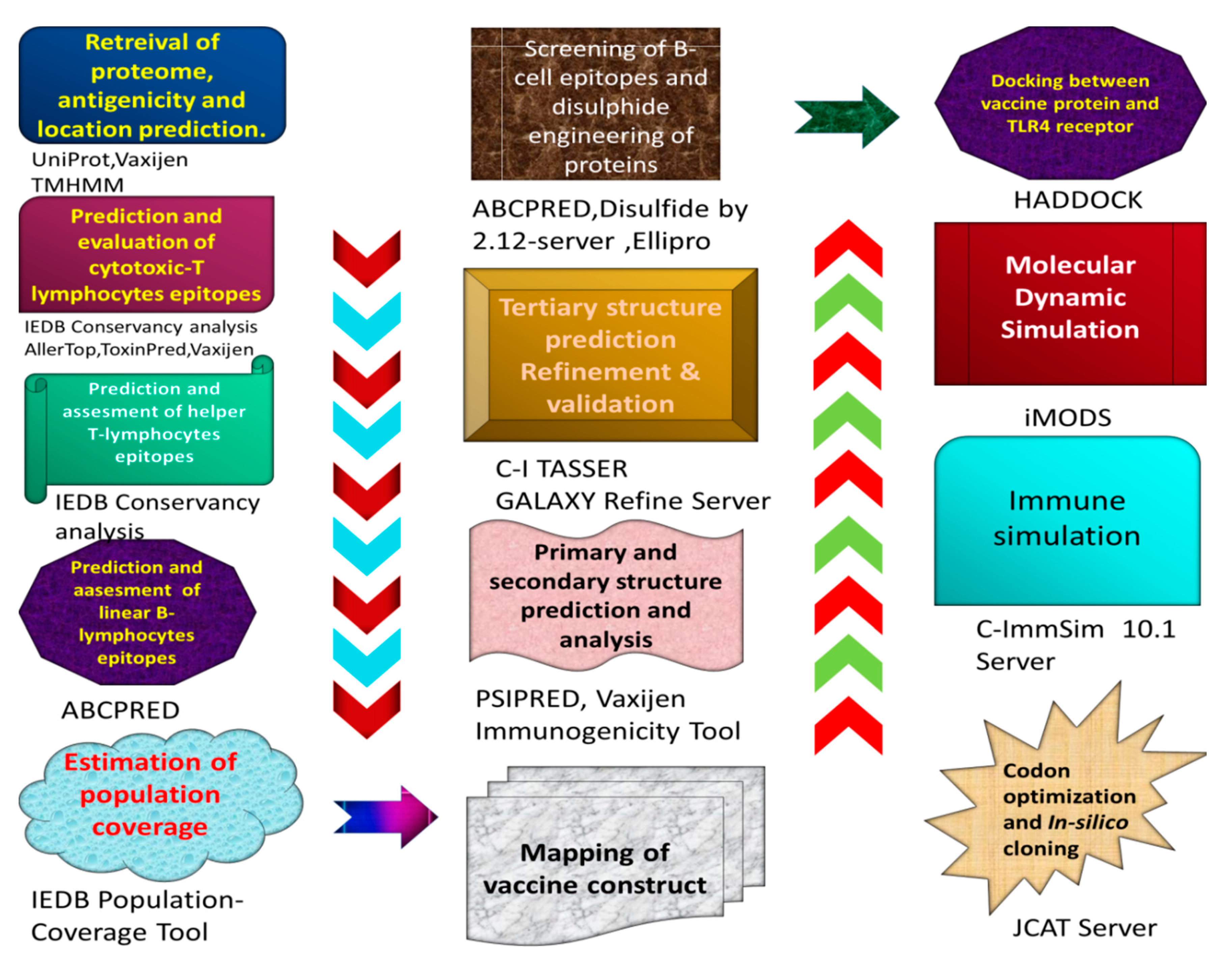
2. Methodology
2.1. Retrieval of Proteom Analysis
2.2. CTL Epitope Selection and Evaluation
2.3. Selection and Analysis of HTL Epitopes
2.4. LBL Epitope Identification and Evaluation
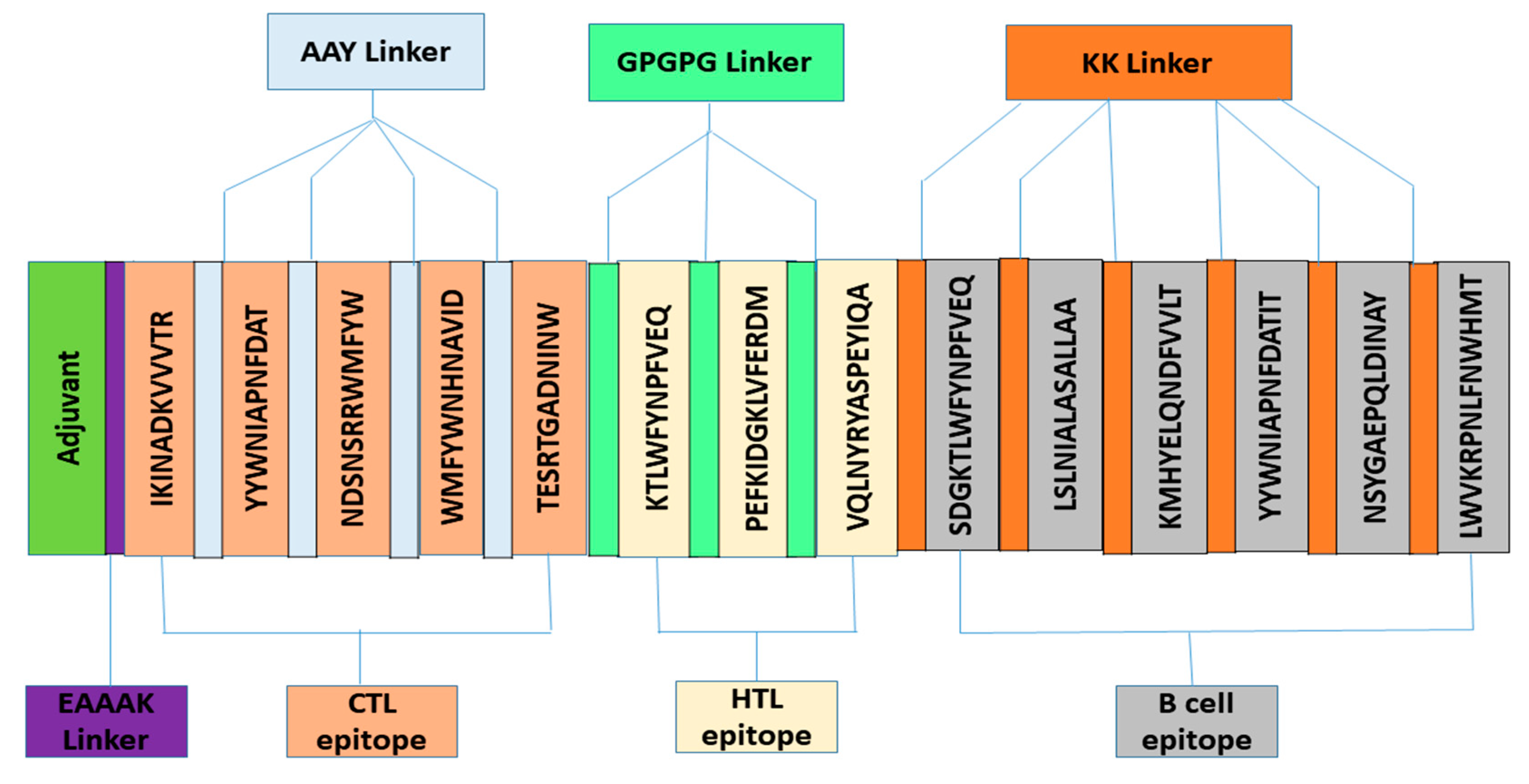
2.5. Vaccine’s Mapping
2.6. Structural Analysis
2.7. Prediction of Tertiary Structure, Confirmation and Refinement
2.8. B-Cell Epitopes Screening
2.9. Disulfide Engineering
2.10. Docking of TLR4 Receptor with Constructed Vaccine Disulfide
2.11. Molecular Dynamic Simulation
2.12. Immunogenicity Evaluation of Construct
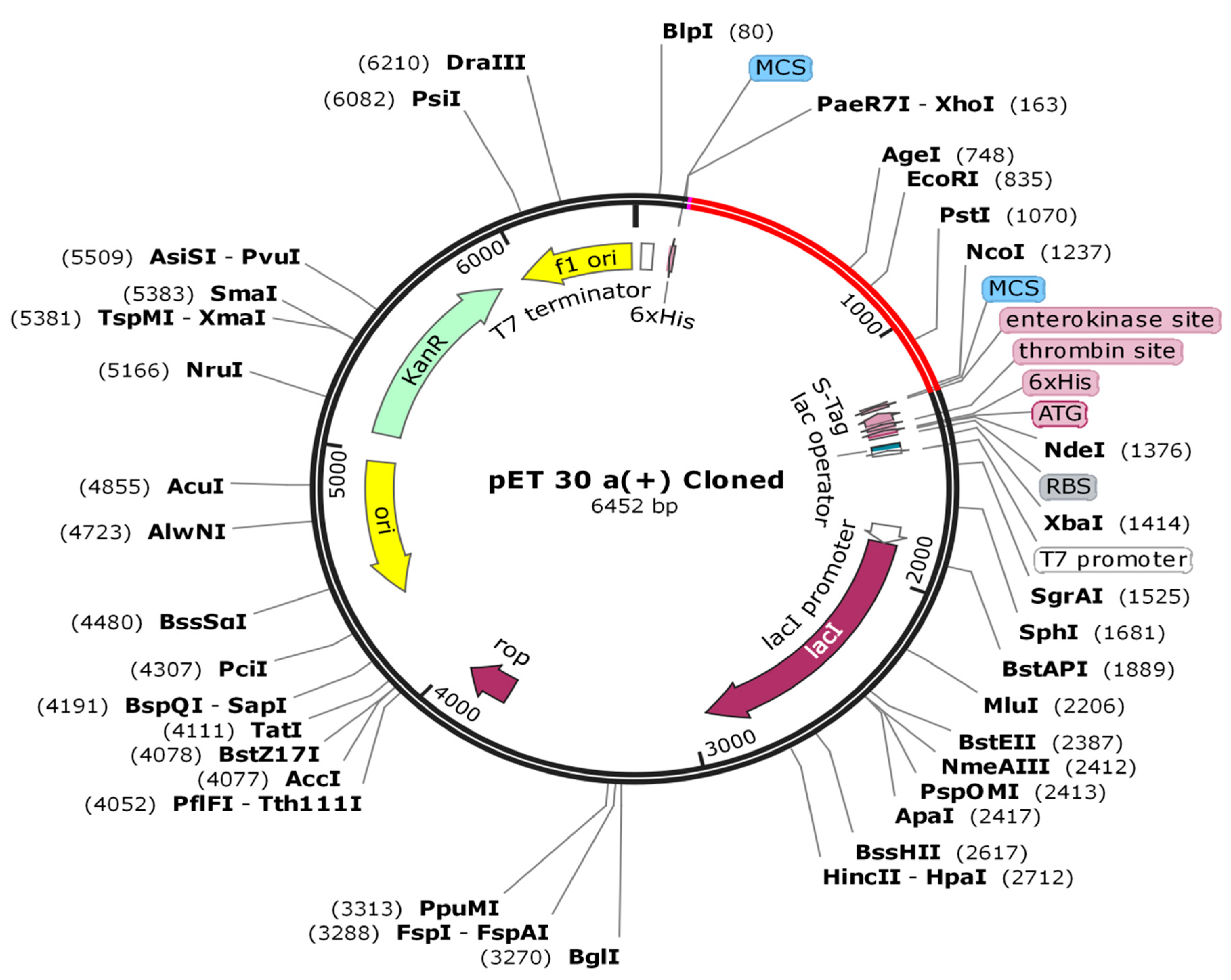
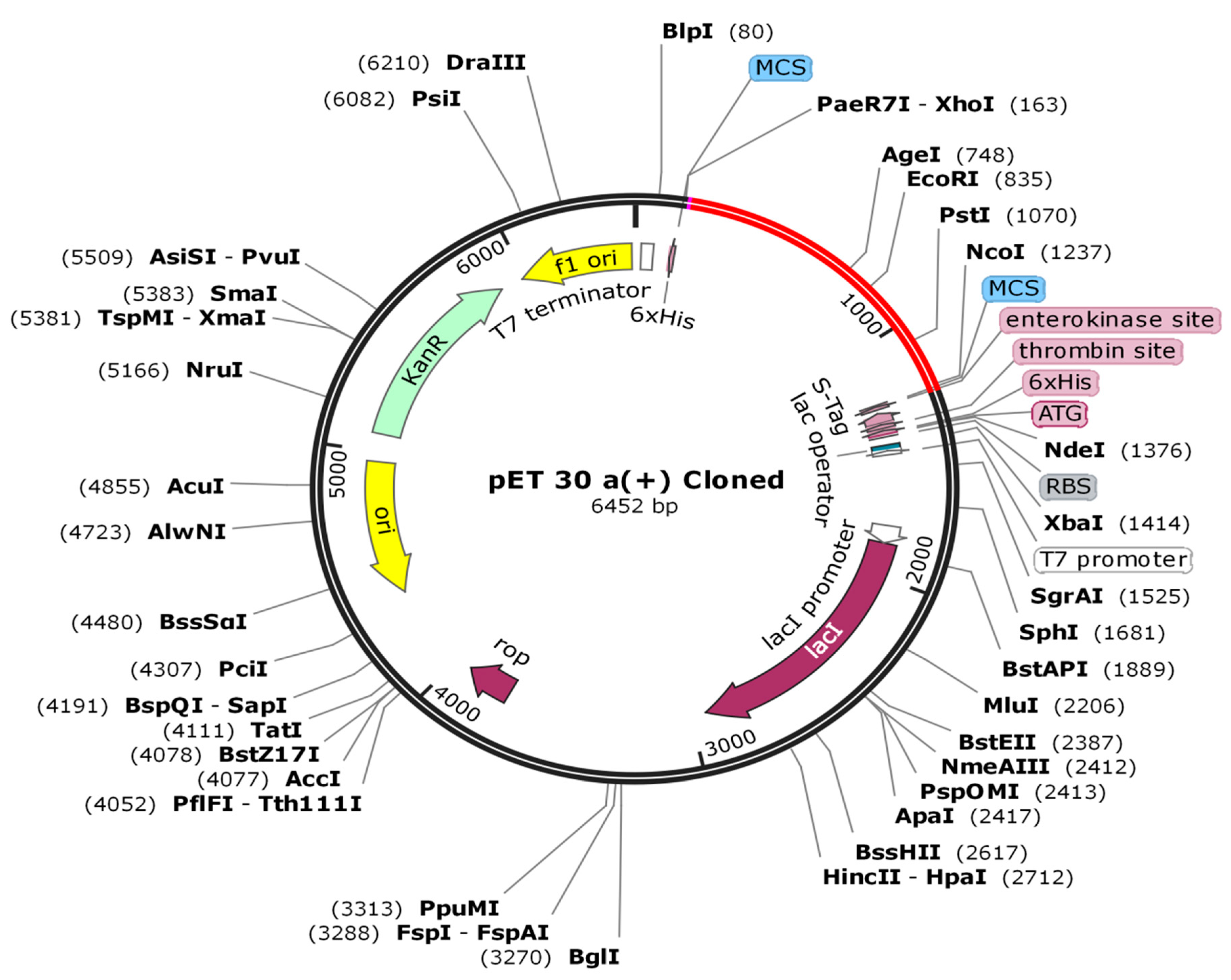
2.13. Codon Adaptation and In Silico Cloning
3. Results
3.1. Protein’s Selection
3.2. CTL Epitope Selection and Evaluation
3.3. Construction of Vaccine
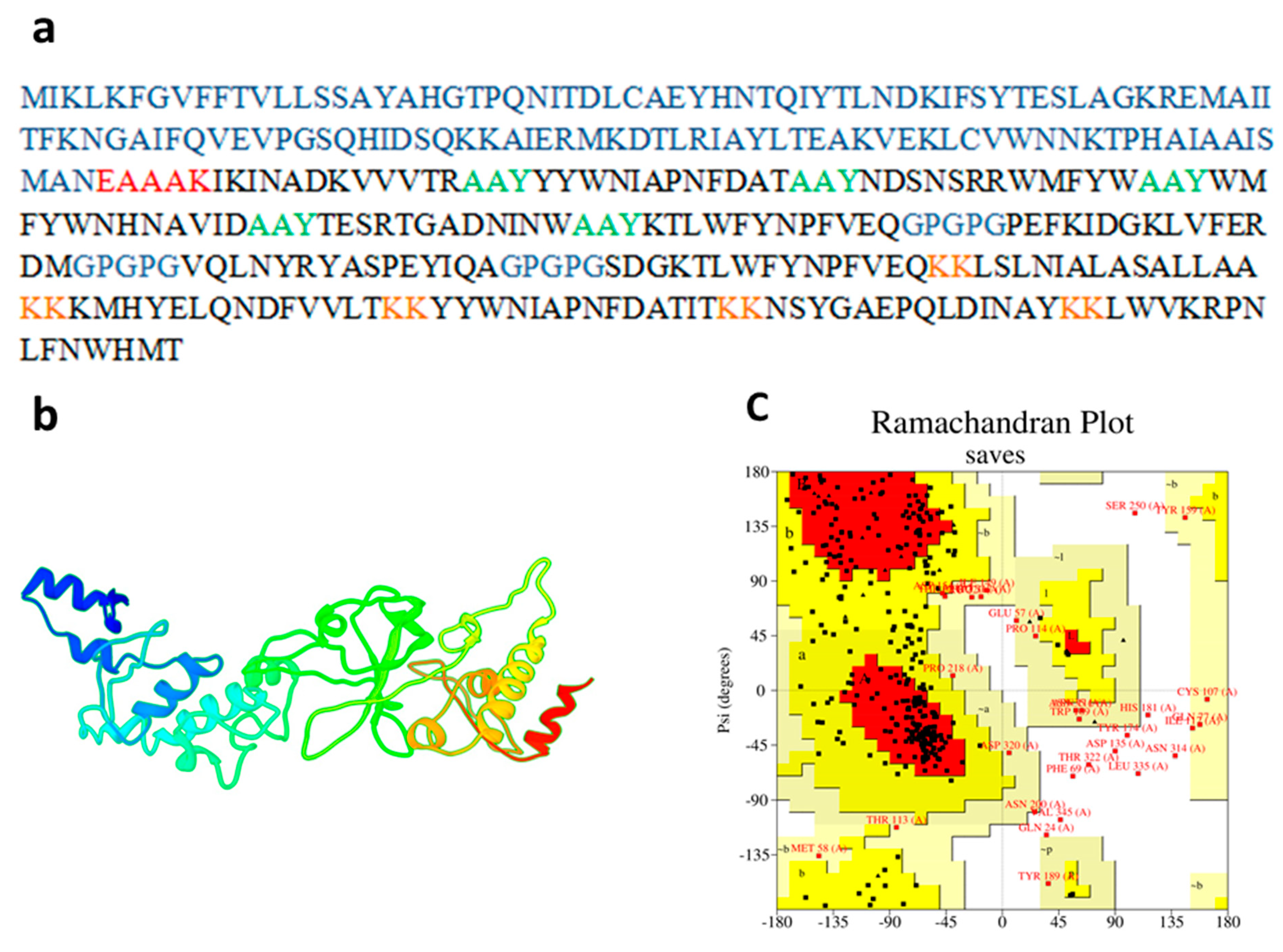
3.4. Physiochemical and Immunogenic Profiling
3.5. Structural Evaluation
3.6. Prediction of Tertiary Structure, Validation and Refinement
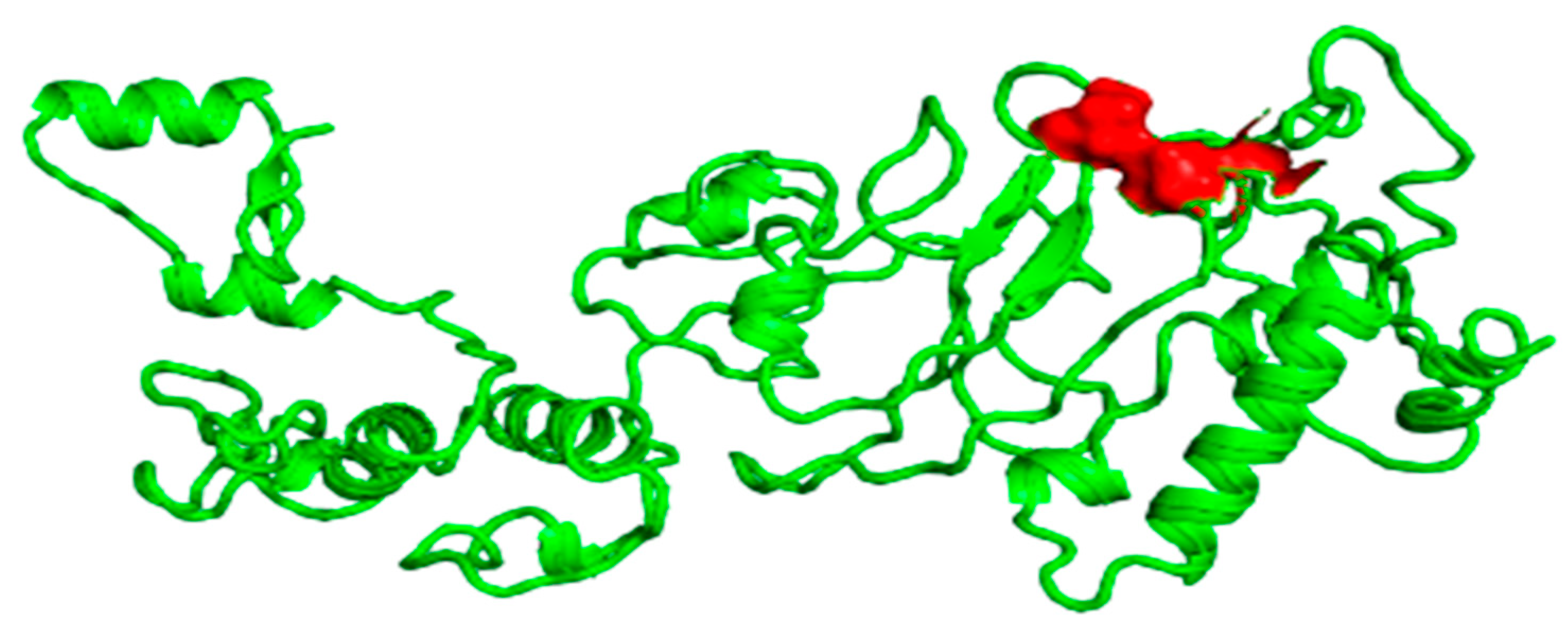
3.7. Selection of B-Cell Epitopes
3.8. Disulfide Engineering
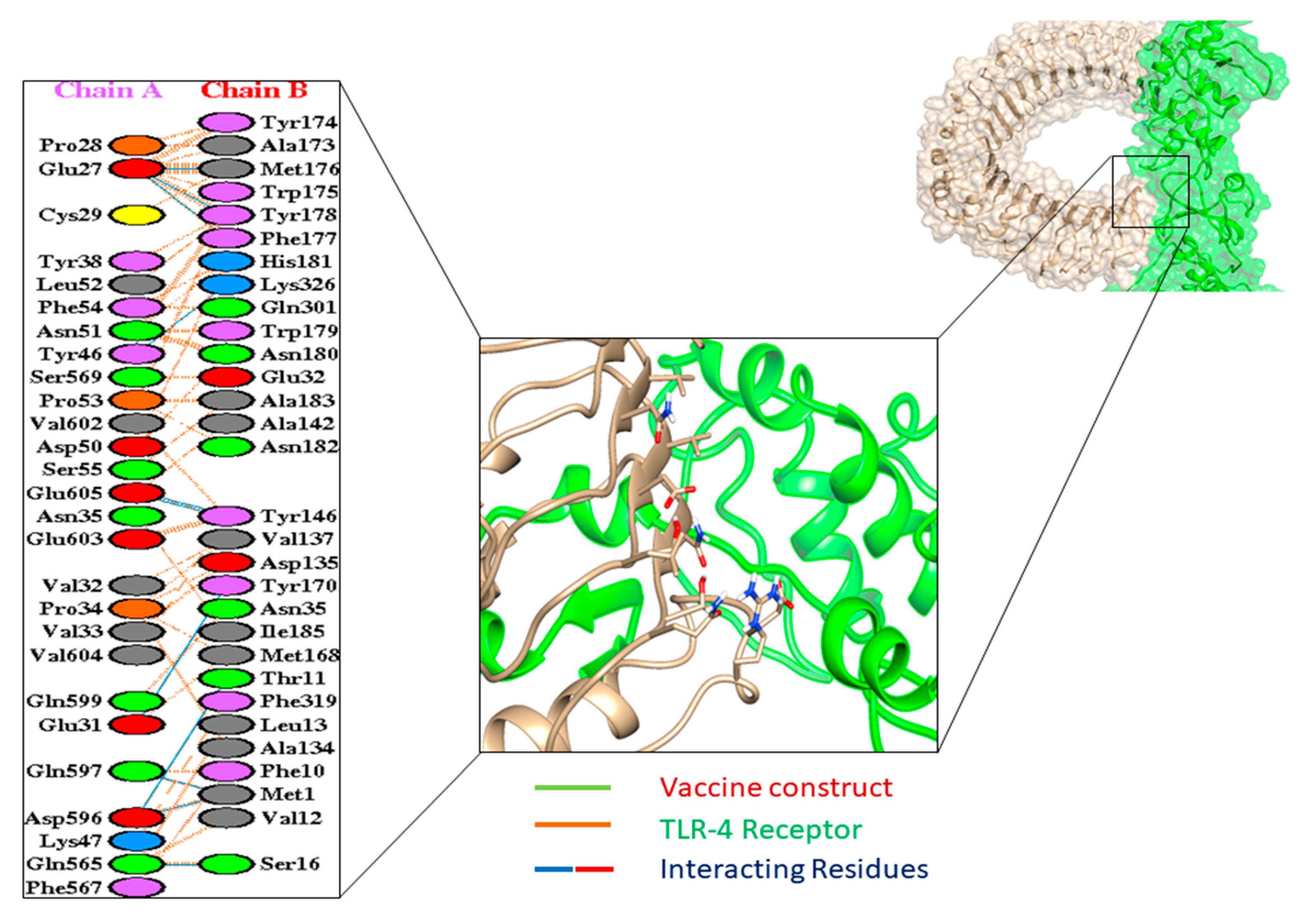
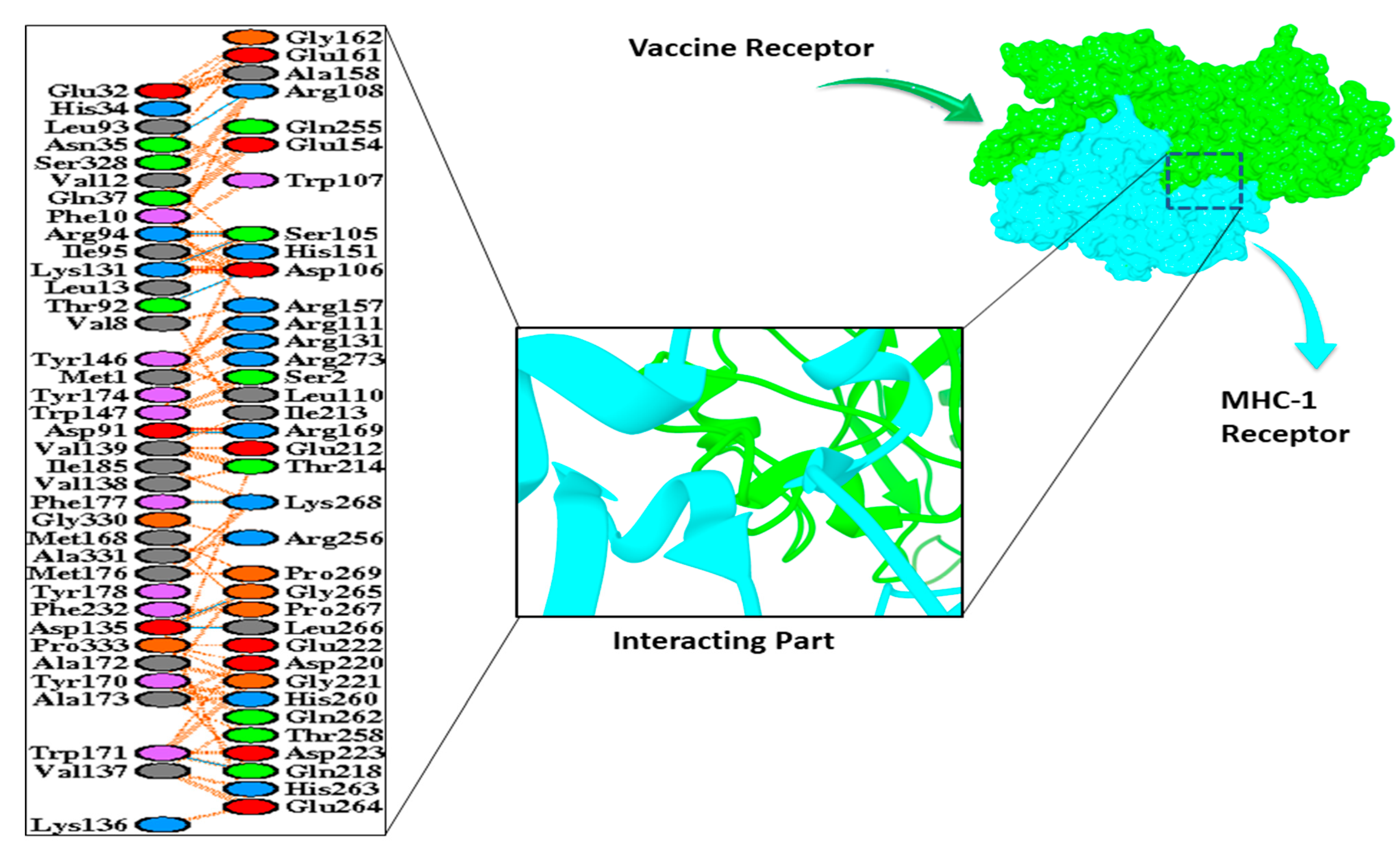
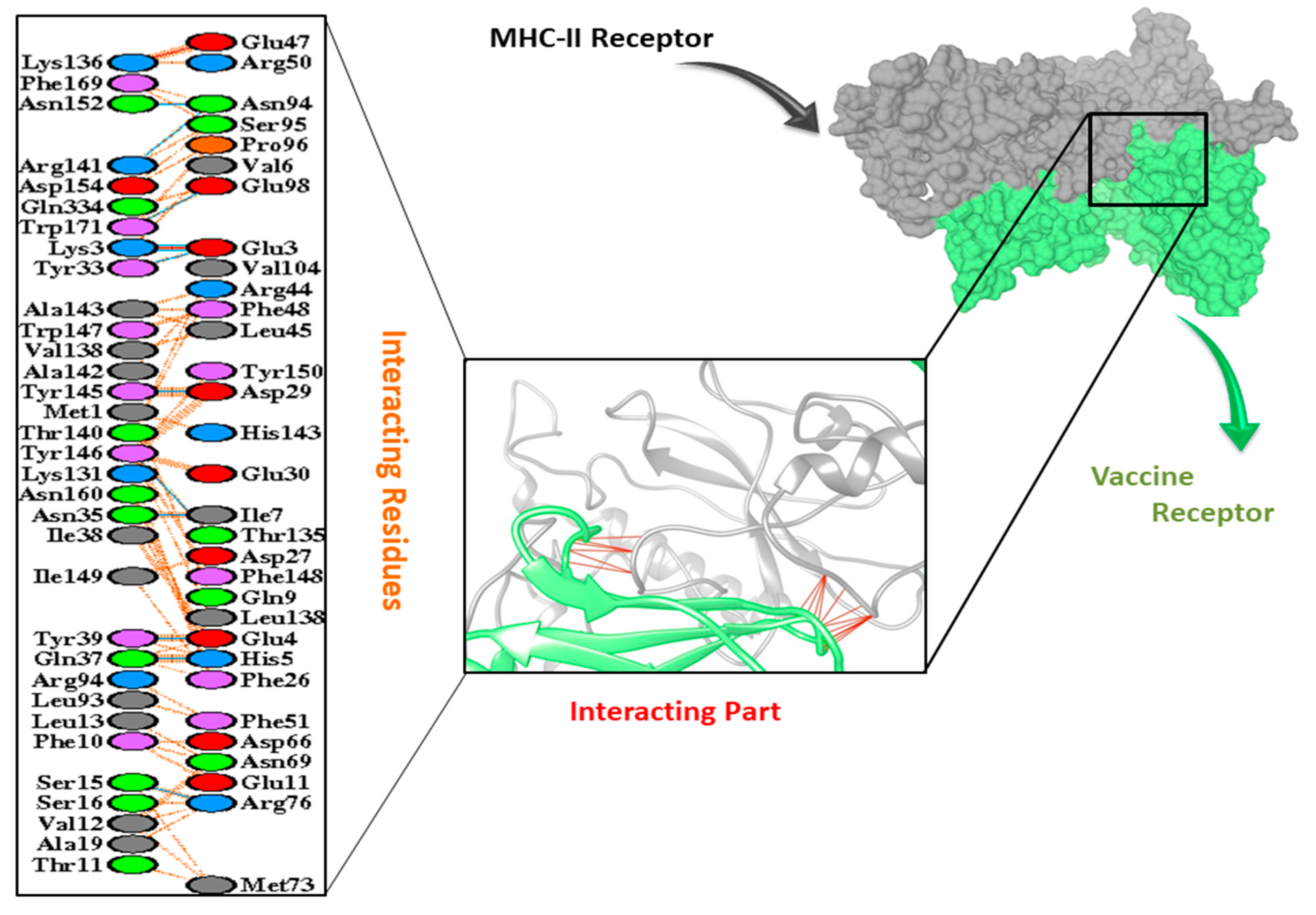
3.9. Molecular Docking
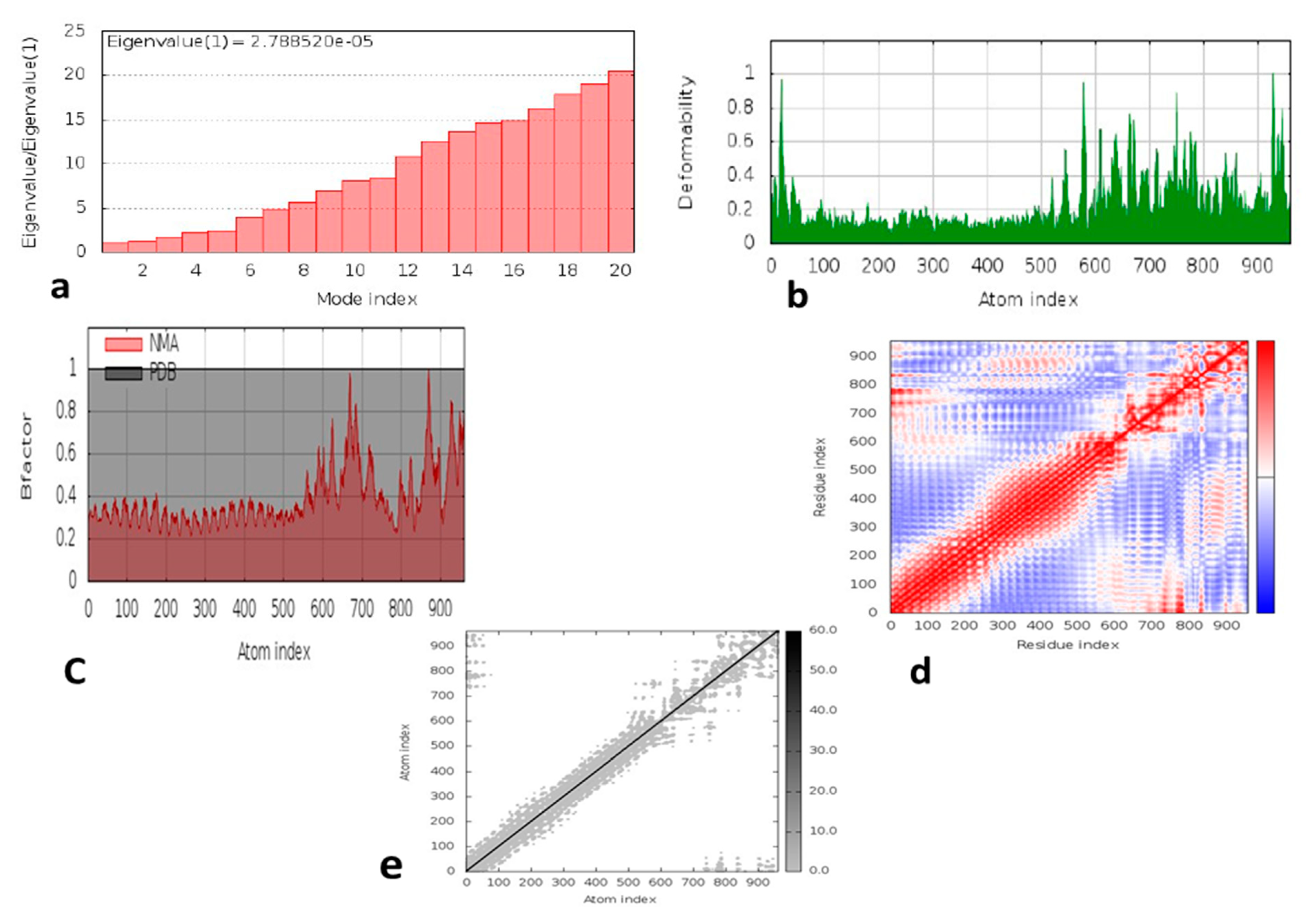
3.10. Molecular Dynamic Simulation
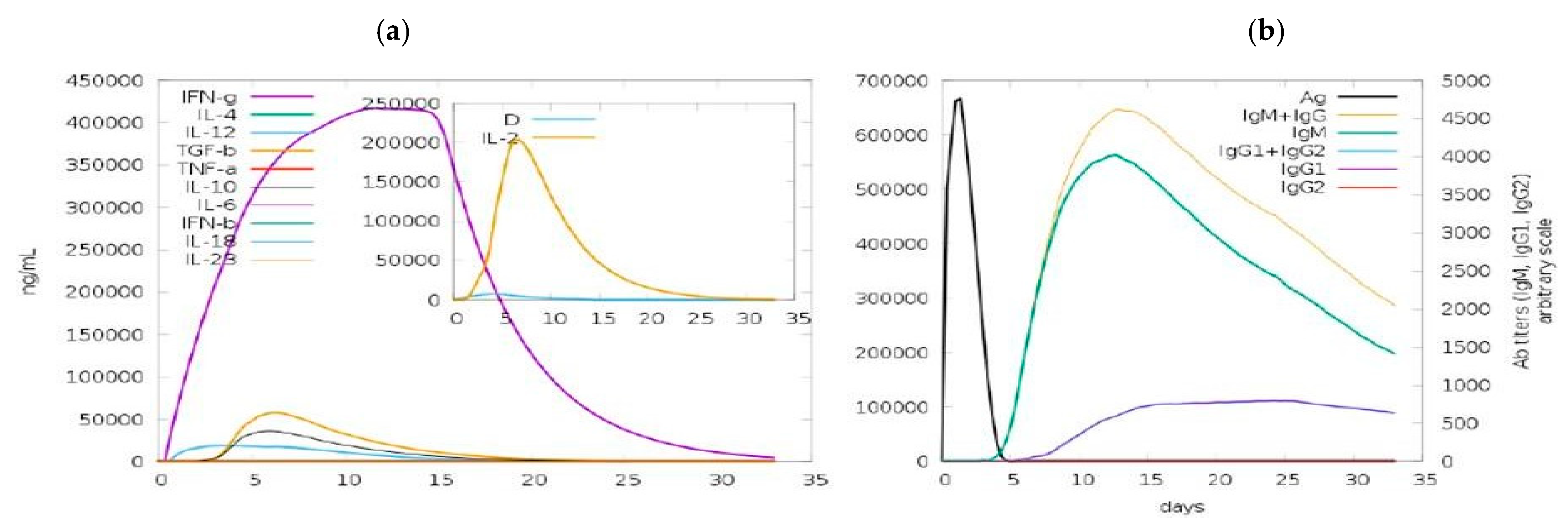
3.11. Immune-Simulation
3.12. In Silico Cloning
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tindall, B.; Sutton, G.; Garrity, G. Enterobacter aerogenes Hormaeche and Edwards 1960 (Approved Lists 1980) and Klebsiella mobilis Bascomb et al. 1971 (Approved Lists 1980) share the same nomenclatural type (ATCC 13048) on the Approved Lists and are homotypic synonyms, with consequences for the name Klebsiella mobilis Bascomb et al. 1971 (Approved Lists 1980). Int. J. Syst. Evol. Microbiol. 2017, 67, 502–504. [Google Scholar] [PubMed]
- Diene, S.M.; Merhej, V.; Henry, M.; El Filali, A.; Roux, V.; Robert, C.; Azza, S.; Gavory, F.; Barbe, V.; La Scola, B.; et al. The rhizome of the multidrug-resistant Enterobacter aerogenes genome reveals how new “killer bugs” are created because of a sympatric lifestyle. Mol. Biol. Evol. 2013, 30, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Davin-Regli, A. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front. Microbiol. 2015, 6, 392. [Google Scholar] [CrossRef] [Green Version]
- Guh, A.Y.; Bulens, S.N.; Mu, Y.; Jacob, J.T.; Reno, J.; Scott, J.; Wilson, L.E.; Vaeth, E.; Lynfield, R.; Shaw, K.M.; et al. Epidemiology of carbapenem-resistant Enterobacteriaceae in 7 US communities, 2012–2013. JAMA 2015, 314, 1479–1487. [Google Scholar] [CrossRef]
- Lee, H.-J.; Choi, J.-K.; Cho, S.-Y.; Kim, S.-H.; Park, S.-H.; Choi, S.-M.; Lee, D.-G.; Choi, J.-H.; Yoo, J.-H. Carbapenem-resistant Enterobacteriaceae: Prevalence and risk factors in a single community-based hospital in Korea. Infect. Chemother. 2016, 48, 166–173. [Google Scholar] [CrossRef]
- Robert, J.; Pantel, A.; Mérens, A.; Lavigne, J.; Nicolas-Chanoine, M. On behalf of ONERBA’s carbapenem resistance study group. Incidence rates of carbapenemase-producing Enterobacteriaceae clinical isolates in France: A prospective nationwide study in 2011–12. J. Antimicrob. Chemother. 2014, 69, 2706–2712. [Google Scholar] [CrossRef] [Green Version]
- Franolić, I.; Bedenić, B.; Beader, N.; Lukić-Grlić, A.; Mihaljević, S.; Bielen, L.; Zarfel, G.; Meštrović, T. NDM-1-producing Enterobacter aerogenes isolated from a patient with a JJ ureteric stent in situ. CEN Case Rep. 2019, 8, 38–41. [Google Scholar] [CrossRef]
- Khajuria, A.; Praharaj, A.K.; Kumar, M.; Grover, N. Carbapenem resistance among Enterobacter species in a tertiary care hospital in central India. Chemother. Res. Pract. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyres, K.L.; Holt, K.E. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 2016, 24, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Simmonds, A.; Annavajhala, M.K.; Wang, Z.; Macesic, N.; Hu, Y.; Giddins, M.J.; O’Malley, A.; Toussaint, N.C.; Whittier, S.; Torres, V.J. Genomic and geographic context for the evolution of high-risk carbapenem-resistant Enterobacter cloacae complex clones ST171 and ST78. MBio 2018, 9, e00542-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Corona, F.; Blanco, P.; Alcalde-Rico, M.; Hernando-Amado, S.; Lira, F.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. The analysis of the antibiotic resistome offers new opportunities for therapeutic intervention. Future Med. Chem. 2016, 8, 1133–1151. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Fajardo, A.; Garmendia, L.; Hernandez, A.; Linares, J.F.; Martínez-Solano, L.; Sánchez, M.B. A global view of antibiotic resistance. FEMS Microbiol. Rev. 2008, 33, 44–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passarelli-Araujo, H.; Palmeiro, J.K.; MOHARANA, K.C.; Pedrosa-Silva, F.; Dalla-Costa, L.M.; Venancio, T.M. Molecular epidemiology of 16S rRNA methyltransferase in Brazil: RmtG in Klebsiella aerogenes ST93 (CC4). Anais Acad. Bras. Ciênc. 2019, 91. [Google Scholar] [CrossRef] [Green Version]
- Grazziotin, A.L.; Vidal, N.M.; Palmeiro, J.K.; Dalla-Costa, L.M.; Venancio, T.M. Genome sequencing of four multidrug-resistant Enterobacter aerogenes isolates from hospitalized patients in Brazil. Front. Microbiol. 2016, 7, 1649. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.-Y.; Huang, H.-Y.; Zou, H.; Wu, M.-L.; Lin, Q.-X.; Liu, B.; Huang, S.-F. Carbapenem-Resistant Klebsiella aerogenes Clinical Isolates from a Teaching Hospital in Southwestern China: Detailed Molecular Epidemiology, Resistance Determinants, Risk Factors and Clinical Outcomes. Infect. Drug Resist. 2020, 13, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, B.; Ashfaq, U.A.; Rahman, M.U.; Masoud, M.S.; Yousaf, M.Z. Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: An immuno-informatics approach. Microb. Pathog. 2019, 132, 243–253. [Google Scholar] [CrossRef]
- Aldakheel, F.M.; Abrar, A.; Munir, S.; Aslam, S.; Allemailem, K.S.; Khurshid, M.; Ashfaq, U.A. Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens. Vaccines 2021, 9, 1079. [Google Scholar] [CrossRef]
- Ashfaq, U.A.; Saleem, S.; Masoud, M.S.; Ahmad, M.; Nahid, N.; Bhatti, R.; Almatroudi, A.; Khurshid, M. Rational design of multi epitope-based subunit vaccine by exploring MERS-COV proteome: Reverse vaccinology and molecular docking approach. PLoS ONE 2021, 16, e0245072. [Google Scholar] [CrossRef]
- Aslam, S.; Ahmad, S.; Noor, F.; Ashfaq, U.A.; Shahid, F.; Rehman, A.; Tahir Ul Qamar, M.; Alatawi, E.A.; Alshabrmi, F.M.; Allemailem, K.S. Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches. Biology 2021, 10, 997. [Google Scholar] [CrossRef]
- Khalid, H.; Ashfaq, U.A. Exploring HCV genome to construct multi-epitope based subunit vaccine to battle HCV infection: Immunoinformatics based approach. J. Biomed. Inform. 2020, 108, 103498. [Google Scholar] [CrossRef]
- Mahmood, M.; Javaid, A.; Shahid, F.; Ashfaq, U.A. Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect. Genet. Evol 2021, 91, 104795. [Google Scholar] [CrossRef]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir Ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Javaid, A.; Khalid, H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect. Genet. Evol 2020, 80, 104199. [Google Scholar] [CrossRef]
- Kalita, P.; Lyngdoh, D.L.; Padhi, A.K.; Shukla, H.; Tripathi, T. Development of multi-epitope driven subunit vaccine against Fasciola gigantica using immunoinformatics approach. Int. J. Biol. Macromol. 2019, 138, 224–233. [Google Scholar] [CrossRef]
- Azhagesan, K.; Ravindran, B.; Raman, K. Network-based features enable prediction of essential genes across diverse organisms. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.; Consortium, O.S.D.D. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood J. Am. Soc. Hematol. 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Gupta, S.; Vir, P.; Raghava, G. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.K.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, 1–10. [Google Scholar]
- Cooper, M.D. The early history of B cells. Nat. Rev. Immunol. 2015, 15, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Nain, Z.; Abdulla, F.; Rahman, M.M.; Karim, M.M.; Khan, M.S.A.; Sayed, S.B.; Mahmud, S.; Rahman, S.R.; Sheam, M.M.; Haque, Z. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Nezafat, N.; Ghasemi, Y.; Javadi, G.; Khoshnoud, M.J.; Omidinia, E. A novel multi-epitope peptide vaccine against cancer: An in silico approach. J. Theor. Biol. 2014, 349, 121–134. [Google Scholar] [CrossRef]
- Mahram, A.; Herbordt, M.C. Fast and accurate NCBI BLASTP: Acceleration with multiphase FPGA-based prefiltering. In Proceedings of the 24th ACM International Conference on Supercomputing, Tsukuba, Japan, 2–4 June 2010; pp. 73–82. [Google Scholar]
- Walker, J.M. The Proteomics Protocols Handbook; Springer: New York, NY, USA, 2005. [Google Scholar]
- Zheng, W.; Li, Y.; Zhang, C.; Pearce, R.; Mortuza, S.; Zhang, Y. Deep-learning contact-map guided protein structure prediction in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1149–1164. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Lengths, M.; Angles, M. Limitations of structure evaluation tools errat. Quick Guidel. Comput. Drug Des. 2018, 16, 75. [Google Scholar]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef]
- Van Aalten, D.M.; De Groot, B.L.; Findlay, J.B.; Berendsen, H.J.; Amadei, A. A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 1997, 18, 169–181. [Google Scholar] [CrossRef]
- Wüthrich, K.; Wagner, G.; Richarz, R.; Braun, W. Correlations between internal mobility and stability of globular proteins. Biophys. J. 1980, 32, 549–560. [Google Scholar] [CrossRef] [Green Version]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PloS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes—Key regulators of immunity. Current Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, J.A.; Chacón, P.; Abagyan, R. Predictions of protein flexibility: First-order measures. Proteins Struct. Funct. Bioinform. 2004, 56, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef]
- Kazi, A.; Chuah, C.; Majeed, A.B.A.; Leow, C.H.; Lim, B.H.; Leow, C.Y. Current progress of immunoinformatics approach harnessed for cellular-and antibody-dependent vaccine design. Pathog. Glob. Health 2018, 112, 123–131. [Google Scholar] [CrossRef]
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing—An overview. Vaccine 2017, 35, 4064–4071. [Google Scholar] [CrossRef]
- Yin, D.; Li, L.; Song, X.; Li, H.; Wang, J.; Ju, W.; Qu, X.; Song, D.; Liu, Y.; Meng, X. A novel multi-epitope recombined protein for diagnosis of human brucellosis. BMC Infect. Dis. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherryholmes, G.A.; Stanton, S.E.; Disis, M.L. Current methods of epitope identification for cancer vaccine design. Vaccine 2015, 33, 7408–7414. [Google Scholar] [CrossRef] [PubMed]
- Zinner, S.; Peter, G. The potential role of cell wall core glycolipids in the immunoprophylaxis and immunotherapy of Gram-negative rod bacteraemia. Med. Microbiol. 1983, 2, 71–85. [Google Scholar]
- Young, S.E. Bacteraemia 1975–1980: A survey of cases reported to the PHLS Communicable Disease Surveillance Centre. J. Infect. 1982, 5, 19–26. [Google Scholar] [CrossRef]
- Cryz, S. Progress in immunization against Klebsiella infections. Eur. J. Clin. Microbiol. 1983, 2, 523–528. [Google Scholar] [CrossRef]
- Ziegler, E.J.; McCutchan, J.A.; Fierer, J.; Glauser, M.P.; Sadoff, J.C.; Douglas, H.; Braude, A.I. Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N. Engl. J. Med. 1982, 307, 1225–1230. [Google Scholar] [CrossRef]
- Lin, X.; Chen, S.; Xue, X.; Lu, L.; Zhu, S.; Li, W.; Chen, X.; Zhong, X.; Jiang, P.; Sename, T.S. Chimerically fused antigen rich of overlapped epitopes from latent membrane protein 2 (LMP2) of Epstein–Barr virus as a potential vaccine and diagnostic agent. Cell. Mol. Immunol. 2016, 13, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.O.; Phanzu, D.M.; Beissner, M.; Badziklou, K.; Luzolo, E.K.; Sarfo, F.S.; Halatoko, W.A.; Amoako, Y.; Frimpong, M.; Kabiru, A.M. Effectiveness of routine BCG vaccination on buruli ulcer disease: A case-control study in the Democratic Republic of Congo, Ghana and Togo. PLoS Negl. Trop. Dis. 2015, 9, e3457. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.J. The enigmatic PE/PPE multigene family of mycobacteria and tuberculosis vaccination. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [Green Version]
- Dheenadhayalan, V.; Delogu, G.; Brennan, M.J. Expression of the PE_PGRS 33 protein in Mycobacterium smegmatis triggers necrosis in macrophages and enhanced mycobacterial survival. Microbes Infect. 2006, 8, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1–11. [Google Scholar]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 1–18. [Google Scholar]
- Hengen, P.N. Purification of His-Tag fusion proteins from Escherichia coli. Trends Biochem. Sci. 1995, 20, 285–286. [Google Scholar] [CrossRef]
- Raines, R.T.; McCormick, M.; Van Oosbree, T.R.; Mierendorf, R.C. [23] The S· tag fusion system for protein purification. Methods Enzymol. 2000, 326, 362–376. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No | Protein Name | Accession No | Antigenicity | Helices | Location |
|---|---|---|---|---|---|
| 1 | Lipopolysaccharide export system protein LptA | A0A0H3FMX7 | 0.77 | 0 | Extracellular |
| 2 | LPS-assembly protein LptD | A0A0H3FNQ6 | 0.67 | 0 | Extracellular |
| 3 | Outer-membrane lipoprotein carrier protein | A0A0H3FTL0 | 0.71 | 0 | Extracellular |
| Epitope | Protein | Alleles | Position | Antigenicity | Immunogenicity |
|---|---|---|---|---|---|
| IKINADKVVVTR | Lipopolysaccharide export system protein LptA | HLA-A*31:01 | 65–76 | 0.9404 | 0.02297 |
| YYWNIAPNFDAT | LPS-assembly protein LptD | HLA-A*23:01 HLA-C*07:02 HLA-C*14:02 HLA-A*24:02 | 247–258 | 1.9021 | 0.36777 |
| NDSNSRRWMFYW | LPS-assembly protein LptD | HLA-B*57:01 HLA-B*58:01 HLA-A*01:01 | 304–315 | 1.8656 | 0.02653 |
| WMFYWNHNAVID | LPS-assembly protein LptD | HLA-C*07:02 HLA-B*39:01 HLA-B*48:01 | 311–322 | 1.3380 | 0.41831 |
| TESRTGADNINW | LPS-assembly protein LptD | HLA-B*44:02 HLA-B*44:03 HLA-B*57:01 | 583–594 | 1.3921 | 0.23133 |
| KTLWFYNPFVEQ | Outer-membrane lipoprotein carrier protein | HLA-A*02:01 HLA-A*32:01 | 85–96 | 0.5371 | 0.51613 |
| Epitope | Protein | Alleles | Position | Antigenicity | IFN-Y | IL-4 | IL-10 |
|---|---|---|---|---|---|---|---|
| PEFKIDGKLVFERDM | LPS-assembly protein LptD | HLA-DRB1*03:09 HLA-DRB1*03:05 HLA-DRB1*03:01 HLA-DRB1*03:06 HLA-DRB1*03:07 HLA-DRB1*03:08 HLA-DRB1*11:28 HLA-DRB1*13:05 HLA-DRB1*11:07 | 483–497 | 0.9915 | Positive | Inducer | Negative |
| VQLNYRYASPEYIQA | LPS-assembly protein LptD | HLA-DRB1*09:01 HLA-DRB1*07:03 HLA-DRB1*11:02 HLA-DRB1*11:21 HLA-DRB1*13:22 | 649–663 | 1.1174 | Positive | Inducer | Negative |
| SDGKTLWFYNPFVEQ | Outer-membrane lipoprotein carrier protein | HLA-DQA1*01:01/DQB1*05:01 | 82–96 | 0.6977 | Positive | Inducer | Negative |
| Epitope | Protein | Score | Position | Antigenicity | Immunogenicity |
|---|---|---|---|---|---|
| LSLNIALASALLAA | Lipopolysaccharide export system protein LptA | 8 | 0.58 | 1.0434 | 0.06495 |
| KMHYELQNDFVVLT | Lipopolysaccharide export system protein LptA | 111 | 0.52 | 0.9433 | 0.1772 |
| YYWNIAPNFDATIT | LPS-assembly protein LptD | 247 | 0.85 | 1.7077 | 0.49894 |
| NSYGAEPQLDINAY | LPS-assembly protein LptD | 386 | 0.68 | 1.7424 | 0.19838 |
| LWVKRPNLFNWHMT | Outer-membrane lipoprotein carrier protein | 60 | 0.68 | 1.1223 | 0.05981 |
| Parameters | Values |
|---|---|
| TLR-4 | |
| HADDOCK-v.2.2 score | 88.6 ± 20.9 |
| Cluster size | 8 |
| RMSD from the overall lowest energy structure | 6.4 ± 0.3 |
| Van-der-Waals energy | −107.4 ± 11.7 |
| Electrostatic energy | −129.9 ± 26.6 |
| Desolvation energy | −68.9 ± 5.7 |
| Restraint violation energy | 2909.2 ± 318.20 |
| Buried surface area | 3238.0 ± 127.9 |
| Z-score | −0.1 |
| MHC-1 Receptor | |
| HADDOCK-v.2.4 score | 169.1 ± 20.3 |
| Cluster size | 7 |
| RMSD from the overall lowest energy structure | 18.7 ± 0.0 |
| Van-der-Waals energy | −125.6 ± 10.0 |
| Electrostatic energy | −246.3 ± 39.8 |
| Desolvation energy | −36.3 ± 4.9 |
| Restraint violation energy | 3803.0 ± 191.5 |
| Buried surface area | 4018.7 ± 264.0 |
| Z-score | −1.2 |
| MHC-II Receptor | |
| HADDOCK-v.2.4 score | 131.8 ± 26.7 |
| Cluster size | 4 |
| RMSD from the overall lowest energy structure | 16.3 ± 0.2 |
| Van-der-Waals energy | −94.5 ± 15.4 |
| Electrostatic energy | −301.3 ± 34.0 |
| Desolvation energy | −53.6 ± 4.1 |
| Restraint violation energy | 3401.9 ± 175.4 |
| Buried surface area | 3492.0 ± 266.7 |
| Z-score | −1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umar, A.; Haque, A.; Alghamdi, Y.S.; Mashraqi, M.M.; Rehman, A.; Shahid, F.; Khurshid, M.; Ashfaq, U.A. Development of a Candidate Multi-Epitope Subunit Vaccine against Klebsiella aerogenes: Subtractive Proteomics and Immuno-Informatics Approach. Vaccines 2021, 9, 1373. https://doi.org/10.3390/vaccines9111373
Umar A, Haque A, Alghamdi YS, Mashraqi MM, Rehman A, Shahid F, Khurshid M, Ashfaq UA. Development of a Candidate Multi-Epitope Subunit Vaccine against Klebsiella aerogenes: Subtractive Proteomics and Immuno-Informatics Approach. Vaccines. 2021; 9(11):1373. https://doi.org/10.3390/vaccines9111373
Chicago/Turabian StyleUmar, Ahitsham, Asma Haque, Youssef Saeed Alghamdi, Mutaib M Mashraqi, Abdur Rehman, Farah Shahid, Mohsin Khurshid, and Usman Ali Ashfaq. 2021. "Development of a Candidate Multi-Epitope Subunit Vaccine against Klebsiella aerogenes: Subtractive Proteomics and Immuno-Informatics Approach" Vaccines 9, no. 11: 1373. https://doi.org/10.3390/vaccines9111373
APA StyleUmar, A., Haque, A., Alghamdi, Y. S., Mashraqi, M. M., Rehman, A., Shahid, F., Khurshid, M., & Ashfaq, U. A. (2021). Development of a Candidate Multi-Epitope Subunit Vaccine against Klebsiella aerogenes: Subtractive Proteomics and Immuno-Informatics Approach. Vaccines, 9(11), 1373. https://doi.org/10.3390/vaccines9111373