
Advancements in mRNA Encoded Antibodies for Passive Immunotherapy

Abstract
:1. Introduction
2. mRNA as a Platform for Efficient Protein Expression In Vivo
3. Modified mRNA
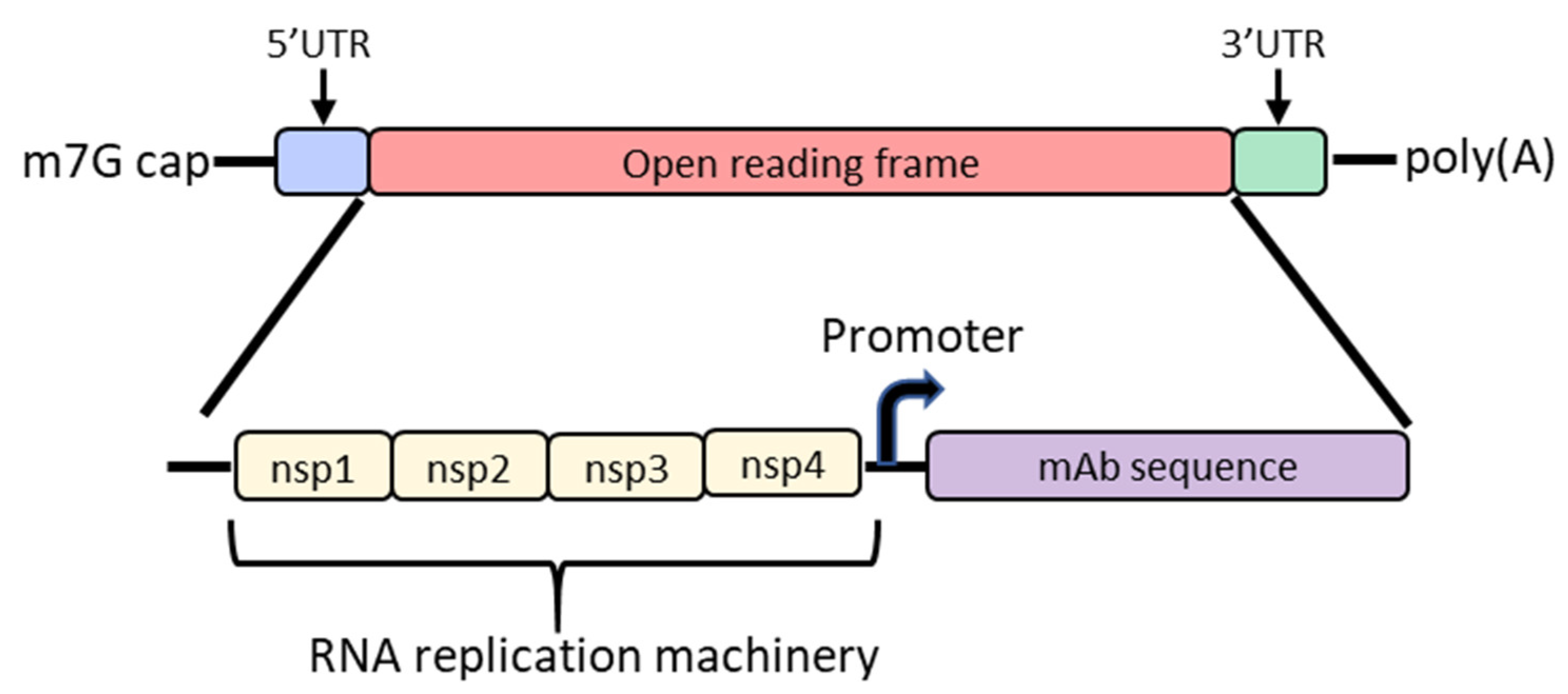
4. Self-Amplifying mRNA
5. mRNA Delivery with LNPs
6. mRNA/LNP Mediated In Vivo Antibody Expression
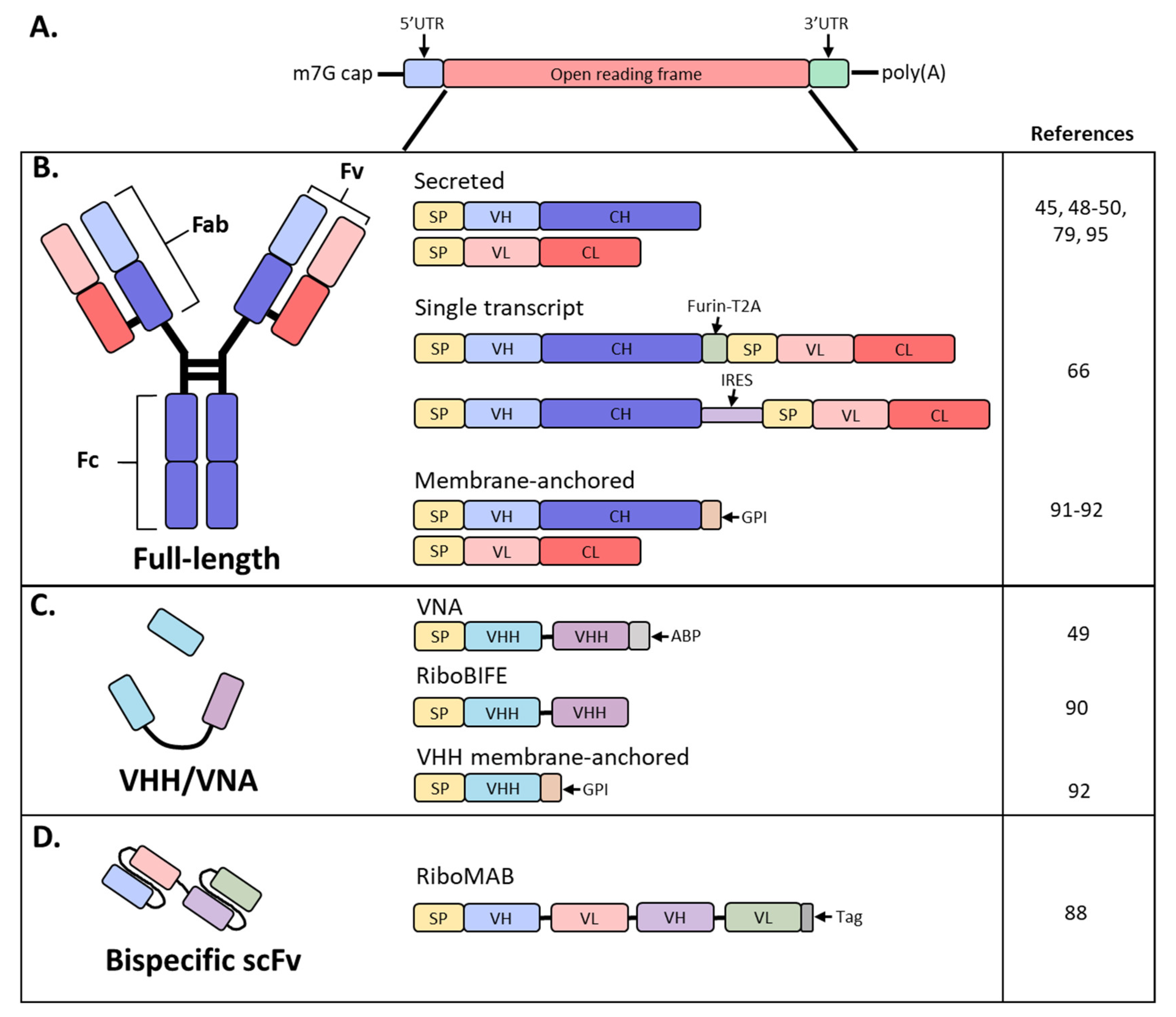
7. Full Length Antibodies
8. Single-Chain Antibodies
9. Engineering mRNA Antibody for Local Delivery
10. mRNA-Encoded Antibodies in Clinical Trials
11. Future of mRNA-Encoded Antibodies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plotkin, S.A. Correlates of Protection Induced by Vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, G. Therapeutic antibodies—Delivering the promise? Adv. Drug Deliver. Rev. 2006, 58, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 Critically Ill Patients with COVID-19 with Convalescent Plasma. JAMA 2020, 323, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, E.; Torvaldsen, S.; Newall, A.T.; Wood, J.G.; Sheikh, M.; Kieny, M.P.; Abela-Ridder, B. Recent advances in the development of monoclonal antibodies for rabies post exposure prophylaxis: A review of the current status of the clinical development pipeline. Vaccine 2018, 37, A132–A139. [Google Scholar] [CrossRef] [PubMed]
- Chippaux, J.-P.; Boyer, L.V.; Alagón, A. Post-exposure treatment of Ebola virus using passive immunotherapy: Proposal for a new strategy. J. Venom. Anim. Toxins 2015, 21, 3. [Google Scholar] [CrossRef] [Green Version]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MABs 2019, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MABs 2015, 7, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Tiller, T.; Meffre, E.; Yurasov, S.; Tsuiji, M.; Nussenzweig, M.C.; Wardemann, H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 2008, 329, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- Mkaddem, S.B.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennewein, M.F.; Alter, G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017, 38, 358–372. [Google Scholar] [CrossRef]
- Lu, X.; Machiesky, L.A.; Mel, N.D.; Du, Q.; Xu, W.; Washabaugh, M.; Jiang, X.R.; Wang, J. Characterization of IgG1 Fc Deamidation at Asparagine 325 and Its Impact on Antibody-dependent Cell-mediated Cytotoxicity and FcγRIIIa Binding. Sci. Rep. 2020, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Thomann, M.; Wegele, H.; Avenal, C.; Schlothauer, T.; Gygax, D.; Beck, H. Oxidation of M252 but not M428 in hu-IgG1 is responsible for decreased binding to and activation of hu-FcγRIIa (His131). Biologicals 2017, 50, 125–128. [Google Scholar] [CrossRef]
- Dangi, A.K.; Sinha, R.; Dwivedi, S.; Gupta, S.K.; Shukla, P. Cell Line Techniques and Gene Editing Tools for Antibody Production: A Review. Front. Pharmacol. 2018, 9, 630. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Rouet, R.; Lowe, D.; Christ, D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, M.A.; Tseng, C.-M.L.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Räty, J.K.; Ylä-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pegu, A.; Rao, E.; Doria-Rose, N.; Beninga, J.; McKee, K.; Lord, D.M.; Wei, R.R.; Deng, G.; Louder, M.; et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 2017, 358, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deal, C.E.; Balazs, A.B. Engineering humoral immunity as prophylaxis or therapy. Curr. Opin. Immunol. 2015, 35, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balazs, A.B.; Chen, J.; Hong, C.M.; Rao, D.S.; Yang, L.; Baltimore, D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2012, 481, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, B.P.; Hackett, N.R.; Crystal, R.G.; Boyer, J.L. Rapid/Sustained Anti-anthrax Passive Immunity Mediated by Co-administration of Ad/AAV. Mol. Ther. 2008, 16, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Priddy, F.H.; Lewis, D.J.M.; Gelderblom, H.C.; Hassanin, H.; Streatfield, C.; LaBranche, C.; Hare, J.; Cox, J.H.; Dally, L.; Bendel, D.; et al. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: A phase 1 randomised controlled trial. Lancet HIV 2019, 6, e230–e239. [Google Scholar] [CrossRef] [Green Version]
- Casazza, J.P.; Narpala, S.; Novik, L.; Yamshchikov, G.; Cale, E.; Doria-Rose, N.; Lin, B.C.; McDermott, A.B.; Roederer, M.; Balazs, A.B.; et al. Durable HIV-1 Antibody Production in Humans After AAV8-Mediated Gene Transfer. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI) 2020, Boston, MA, USA, 8–11 March 2020. [Google Scholar]
- Alvarez, R.D.; Barnes, M.N.; Gomez-Navarro, J.; Wang, M.; Strong, T.V.; Arafat, W.; Arani, R.B.; Johnson, M.R.; Roberts, B.L.; Siegal, G.P.; et al. A Cancer Gene Therapy Approach Utilizing an Anti-erbB-2 Single- Chain Antibody-encoding Adenovirus (AD21): A Phase I Trial1. Clin. Cancer Res. 2000, 6, 3081–3087. [Google Scholar]
- Nidetz, N.F.; McGee, M.C.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol. Ther. 2019, 207, 107453. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors. Hum. Vaccines Immunother. 2015, 10, 2875–2884. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, L.H.; Wang, L.; Somanathan, S.; Zhi, Y.; Figueredo, J.; Calcedo, R.; Sanmiguel, J.; Desai, R.A.; Chen, C.S.; Johnston, J.; et al. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat. Med. 2006, 12, 967–971. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Q.; Calcedo, R.; Mays, L.; Bell, P.; Wang, L.; Vandenberghe, L.H.; Grant, R.; Sanmiguel, J.; Furth, E.E.; et al. Adeno-Associated Virus-Mediated Gene Transfer to Nonhuman Primate Liver Can Elicit Destructive Transgene-Specific T Cell Responses. Hum. Gene Ther. 2009, 20, 930–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Lebherz, C.; Weiner, D.J.; Grant, R.; Calcedo, R.; McCullough, B.; Bagg, A.; Zhang, Y.; Wilson, J.M. Erythropoietin gene therapy leads to autoimmune anemia in macaques. Blood 2004, 103, 3300–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöne, D.; Hrycak, C.P.; Windmann, S.; Lapuente, D.; Dittmer, U.; Tenbusch, M.; Bayer, W. Immunodominance of Adenovirus-Derived CD8+ T Cell Epitopes Interferes with the Induction of Transgene-Specific Immunity in Adenovirus-Based Immunization. J. Virol. 2017, 91, e01184-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W. AAV Vectors, Insertional Mutagenesis, and Cancer. Mol. Ther. 2007, 15, 1740–1743. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009, 4, 442–447. [Google Scholar]
- Tjelle, T.E.; Salte, R.; Mathiesen, I.; Kjeken, R. A novel electroporation device for gene delivery in large animals and humans. Vaccine 2006, 24, 4667–4670. [Google Scholar] [CrossRef]
- Somiari, S.; Glasspool-Malone, J.; Drabick, J.J.; Gilbert, R.A.; Heller, R.; Jaroszeski, M.J.; Malone, R.W. Theory and in Vivo Application of Electroporative Gene Delivery. Mol. Ther. 2000, 2, 178–187. [Google Scholar] [CrossRef]
- Molnar, M.J.; Gilbert, R.; Lu, Y.; Liu, A.-B.; Guo, A.; Larochelle, N.; Orlopp, K.; Lochmuller, H.; Petrof, B.J.; Nalbantoglu, J.; et al. Factors Influencing the Efficacy, Longevity, and Safety of Electroporation-Assisted Plasmid-Based Gene Transfer into Mouse Muscles. Mol Ther. 2004, 10, 447–455. [Google Scholar] [CrossRef]
- Tjelle, T.E.; Corthay, A.; Lunde, E.; Sandlie, I.; Michaelsen, T.E.; Mathiesen, I.; Bogen, B. Monoclonal Antibodies Produced by Muscle after Plasmid Injection and Electroporation. Mol. Ther. 2004, 9, 328–336. [Google Scholar] [CrossRef]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Khoshnejad, M.; Patel, A.; Wojtak, K.; Kudchodkar, S.B.; Humeau, L.; Lyssenko, N.N.; Rader, D.J.; Muthumani, K.; Weiner, D.B. Development of Novel DNA-Encoded PCSK9 Monoclonal Antibodies as Lipid-Lowering Therapeutics. Mol. Ther. 2019, 27, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.-G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef]
- Jain, R.; Frederick, J.P.; Huang, E.Y.; Burke, K.E.; Mauger, D.M.; Andrianova, E.A.; Farlow, S.J.; Siddiqui, S.; Pimentel, J.; Cheung-Ong, K.; et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic Acid Ther. 2018, 28, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Shuman, S. Catalytic Activity of Vaccinia mRNA Capping Enzyme Subunits Coexpressed in Escherichia coZi*. J. Biol. Chem. 1990, 265, 11960–11966. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Guhaniyogi, J.; Brewer, G. Regulation of mRNA stability in mammalian cells. Gene 2001, 265, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-based therapeutic gene delivery by expression augmenting 3’-untranslated regions identified by cellular library screening. Mol. Ther. 2018, 27, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [Green Version]
- Wolff, J.; Malone, R.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Alonso, F.J.; Dabrowski, M.; Wadzack, J.; Nierhaus, K.H. Self-coded 3′-Extension of Run-off Transcripts Produces Aberrant Products during in Vitro Transcription with T7 RNA Polymerase. J. Biol. Chem. 1995, 270, 6298–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholamalipour, Y.; Karunanayake Mudiyanselage, A.; Martin, C.T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, W.; Durnell, L.A.; et al. Intramuscular delivery of replicon RNA encoding ZIKV-117 human monoclonal antibody protects against Zika virus infection. Mol. Ther.-Methods Clin. Dev. 2020, 18, 402–414. [Google Scholar] [CrossRef]
- Melton, D.A.; Krieg, P.A.; Rebagliati, M.R.; Maniatis, T.; Zinn, K.; Green, M.R. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984, 12, 7035–7056. [Google Scholar] [CrossRef] [Green Version]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2020, 1–13. [Google Scholar] [CrossRef]
- Krieg, P.A. Improved synthesis of full-length RNA probe at reduced incubation temperatures. Nucleic Acids Res. 1990, 18, 6463. [Google Scholar] [CrossRef] [Green Version]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, O.; et al. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol. Ther. 2019, 28, 119–128. [Google Scholar] [CrossRef]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.R.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef]
- Diken, M.; Kreiter, S.; Selmi, A.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011, 18, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Hoecke, L.V.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [Green Version]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. Biodrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery For Therapeutic Anti-Her2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Kozma, G.T.; Mészáros, T.; Vashegyi, I.; Fülöp, T.; Örfi, E.; Dézsi, L.; Rosivall, L.; Bavli, Y.; Urbanics, R.; Mollnes, T.E.; et al. Pseudo-anaphylaxis to Polyethylene Glycol (PEG)-Coated Liposomes: Roles of Anti-PEG IgM and Complement Activation in a Porcine Model of Human Infusion Reactions. ACS Nano 2019, 13, 9315–9324. [Google Scholar] [CrossRef] [Green Version]
- Sapparapu, G.; Fernandez, E.; Kose, N.; Cao, B.; Fox, J.M.; Bombardi, R.G.; Zhao, H.; Nelson, C.A.; Bryan, A.L.; Barnes, T.; et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016, 540, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.C.L.; Bardor, M.; Li, B.; Lee, J.J.; Song, Z.; Tong, Y.W.; Goh, L.T.; Yang, Y. Comparison of Internal Ribosome Entry Site (IRES) and Furin-2A (F2A) for Monoclonal Antibody Expression Level and Quality in CHO Cells. PLoS ONE 2013, 8, e63247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, J.; Wang, T.; Nian, R.; Lau, A.; Hoi, K.M.; Ho, S.C.; Gagnon, P.; Bi, X.; Yang, Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MABs 2015, 7, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Welles, H.C.; Jennewein, M.F.; Mason, R.D.; Narpala, S.; Wang, L.; Cheng, C.; Zhang, Y.; Todd, J.P.; Lifson, J.D.; Balazs, A.B.; et al. Vectored delivery of anti-SIV envelope targeting mAb via AAV8 protects rhesus macaques from repeated limiting dose intrarectal swarm SIVsmE660 challenge. PLoS Pathog. 2018, 14, e1007395. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Tremblay, J.M.; Leysath, C.E.; Ofori, K.; Baldwin, K.; Feng, X.; Bedenice, D.; Webb, R.P.; Wright, P.M.; Smith, L.A.; et al. A Novel Strategy for Development of Recombinant Antitoxin Therapeutics Tested in a Mouse Botulism Model. PLoS ONE 2012, 7, e29941. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, J.M.; Mukherjee, J.; Leysath, C.E.; Debatis, M.; Ofori, K.; Baldwin, K.; Boucher, C.; Peters, R.; Beamer, G.; Sheoran, A.; et al. A Single VHH-Based Toxin-Neutralizing Agent and an Effector Antibody Protect Mice against Challenge with Shiga Toxins 1 and 2. Infect. Immun. 2013, 81, 4592–4603. [Google Scholar] [CrossRef] [Green Version]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef]
- Vlieger, D.D.; Hoffmann, K.; Molle, I.V.; Nerinckx, W.; Hoecke, L.V.; Ballegeer, M.; Creytens, S.; Remaut, H.; Hengel, H.; Schepens, B.; et al. Selective Engagement of FcγRIV by a M2e-Specific Single Domain Antibody Construct Protects Against Influenza A Virus Infection. Front. Immunol. 2019, 10, 2920. [Google Scholar] [CrossRef] [Green Version]
- Hoecke, L.V.; Verbeke, R.; Vlieger, D.D.; Dewitte, H.; Roose, K.; Nevel, S.V.; Krysko, O.; Bachert, C.; Schepends, B.; Lentacker, I.; et al. mRNA encoding a bispecific single domain antibody construct protects against influenza A virus infection in mice. Mol. Ther.-Nucleic Acids 2020, 20, 777–787. [Google Scholar] [CrossRef]
- Lindsay, K.E.; Vanover, D.; Thoresen, M.; King, H.; Xiao, P.; Badial, P.; Araínga, M.; Park, S.B.; Tiwari, P.M.; Peck, H.E.; et al. Aerosol delivery of synthetic mRNA to vaginal mucosa leads to durable expression of broadly neutralizing antibodies against HIV. Mol. Ther. 2020, 28, 805–819. [Google Scholar] [CrossRef]
- Tiwari, P.M.; Vanover, D.; Lindsay, K.E.; Bawage, S.S.; Kirschman, J.L.; Bhosle, S.; Lifland, A.W.; Zurla, C.; Santangelo, P.J. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 2018, 9, 3999. [Google Scholar] [CrossRef] [PubMed]
- Resch, B. Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum. Vaccines Immunother. 2017, 13, 2138–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenberg, O.F.; Magnus, C.; Rusert, P.; Günthard, H.F.; Regoes, R.R.; Trkola, A. Predicting HIV-1 transmission and antibody neutralization efficacy in vivo from stoichiometric parameters. PLoS Pathog. 2017, 13, e1006313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaks, T. Antibody against Chikungunya Virus (mRNA-1944) [Powerpoint slides 15-25]. Available online: https://investors.modernatx.com/static-files/8ea64970-8299-43a6-81af-edaf30040fea (accessed on 1 December 2020).
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Delivery Method | Advantages | Disadvantages |
|---|---|---|
| Recombinant protein |
|
|
| Viral vectored |
|
|
| DNA |
|
|
| mRNA |
|
|
| Antibody | Antibody Format (H:L Molar Ratio) | Antigen Target | Specific Modifications | Formulation | Species | Maximum Titer (Dose) | Citation | |
|---|---|---|---|---|---|---|---|---|
| Coexpressed | VRC01 | Full-length (1:1) | HIV (CD4bs) | N1-methyl-pseudouridine | LNP | BALB/c | 170 µg/mL (1.4 mg/kg) | Pardi, N. et al. 2017 |
| BLT mice | 200 µg/mL (1.4 mg/kg) | |||||||
| S057/CR57 | Full-length (1.5:1) | Rabies (glycoprotein G) | Human codon optimization with GC enrichment | LNP | Swiss-Albino mice | 10 µg/mL (2 mg/kg) | Thran, M.; et al. 2017 | |
| CR8033 | Influenza B (HA) | 10 µg/mL (2 mg/kg) | ||||||
| Rituximab | CD20 | NOD/SCID mice | N.D. (2.5 mg/kg) | |||||
| Anti-influenza A human IgG | Full-length (N.D.) | Influenza A | N1-methyl-pseudouridine | LNP | Cynomologous NHP | 4 µg/mL (0.3 mg/kg) | Sabnis, S.; et al. 2018 | |
| CHKV-24 | Full-length (N.D.) | Chikungunya virus | N1-methyl-pseudouridine | LNP | AG129 mice | 14.9 µg/mL (0.5 mg/kg) | Kose, N.; et al. 2019 | |
| Cynomologous NHP | 10.1 µg/mL (0.5 mg/kg) | |||||||
| 1st dose: 16.2 µg/mL (3 mg/kg) 2nd dose: 28.8 µg/mL (3 mg/kg) | ||||||||
| mRNA-1944 | Full-length (N.D.) | Chikungunya virus | N.D. | LNP | Human | 2 µg/mL avg (0.1 mg/kg) | Zaks, T | |
| 7.9 µg/mL avg (0.3 mg/kg) | ||||||||
| 10.2 µg/mL avg (0.6 mg/kg) | ||||||||
| 6.1 µg/mL avg (0.6 mg/kg + steroids) | ||||||||
| 1st dose: 7.2 µg/mL avg (0.3 mg/kg) 2nd dose: 12.9 µg/mL avg (0.3 mg/kg) | ||||||||
| Trastuzumab | Full-length (2:1) | Human Her2 | N.D. | LNP | C57Bl/6 | 57.7 µg/mL (2 mg/kg) | Rybakova, Y.; et al. 2019 | |
| Single-chain antibodies | VNA-BoNTA | Two VHHs fused together with albumin-binding peptide | Botulism toxin A | Human codon optimization with GC enrichment | TransIT | CD1 mice | ~200–400 µg/mL (2 mg/kg) | Thran, M.; et al. 2017 |
| VNA-Stx2 | Two VHHs fused together with albumin-binding peptide | E. coli shiga toxin 2 | ~20–50 µg/mL (2 mg/kg) | |||||
| CD3x tumor associated antigen | RiboMAb: bispecific ScFv | Tumors expressing CLDn6, CLDn18.2 or EpCAM | N1-methyl-pseudouridine | TransIT | NSG mice | 7 µg/mL (0.25 mg/kg) | Stadler, C.R.; et al. 2017 | |
| Self-amplifying mRNA | ZIKV-117 | IRES-linked full length | Zika virus envelope protein | VEEV strain TC-83 nsP1 to nsP4 genes upstream of ZIKV117 open reading frame | NLC | C57Bl/6 | 1.19 µg/mL (2 mg/kg) | Erasmus, J.H.; et al. 2020 |
| Furin-T2A-linked full length | 2.61 µg/mL (2 mg/kg) | |||||||
| Membrane bound/local delivery | Palivizumab | Full-length (4:1) GPI anchor on heavy chain | RSV | N1-methyl-pseudouridine | None | BALB/c | N.D. (5 mg/kg) | Tiwari, P.M.; et al. 2018 |
| RSV aVHH | VHH with GPI anchor | |||||||
| aPGT121 | Full-length (4:1) GPI anchor on heavy chain | HIV env | N1-methyl-pseudouridine | None | Sheep | 210 µg/mL (2 doses of 750 µg each) | Lindsay, K.E.; et al. 2020 | |
| Macaques | N.D. (1000 µg) | |||||||
| sPGT121 | Full-length (4:1) | Sheep | 80 µg/mL (2 doses of 750 µg each) | |||||
| FcγRIV VHH-M2e | RiboBiFE; VHH bispecific Fc-receptor-engaging | Mouse FcγRIV and influenza A M2 extracellular domain | N1-methyl-pseudouridine | LNP | BALB/c | N.D. (0.25 mg/kg) | Hoecke, L.V.; et al. 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deal, C.E.; Carfi, A.; Plante, O.J. Advancements in mRNA Encoded Antibodies for Passive Immunotherapy. Vaccines 2021, 9, 108. https://doi.org/10.3390/vaccines9020108
Deal CE, Carfi A, Plante OJ. Advancements in mRNA Encoded Antibodies for Passive Immunotherapy. Vaccines. 2021; 9(2):108. https://doi.org/10.3390/vaccines9020108
Chicago/Turabian StyleDeal, Cailin E., Andrea Carfi, and Obadiah J. Plante. 2021. "Advancements in mRNA Encoded Antibodies for Passive Immunotherapy" Vaccines 9, no. 2: 108. https://doi.org/10.3390/vaccines9020108