
Quality Verification with a Cluster−Controlled Manufacturing System to Generate Monocyte−Derived Dendritic Cells

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Ethics Statement
2.2. DC Generation
2.3. Morphological Cell Analysis
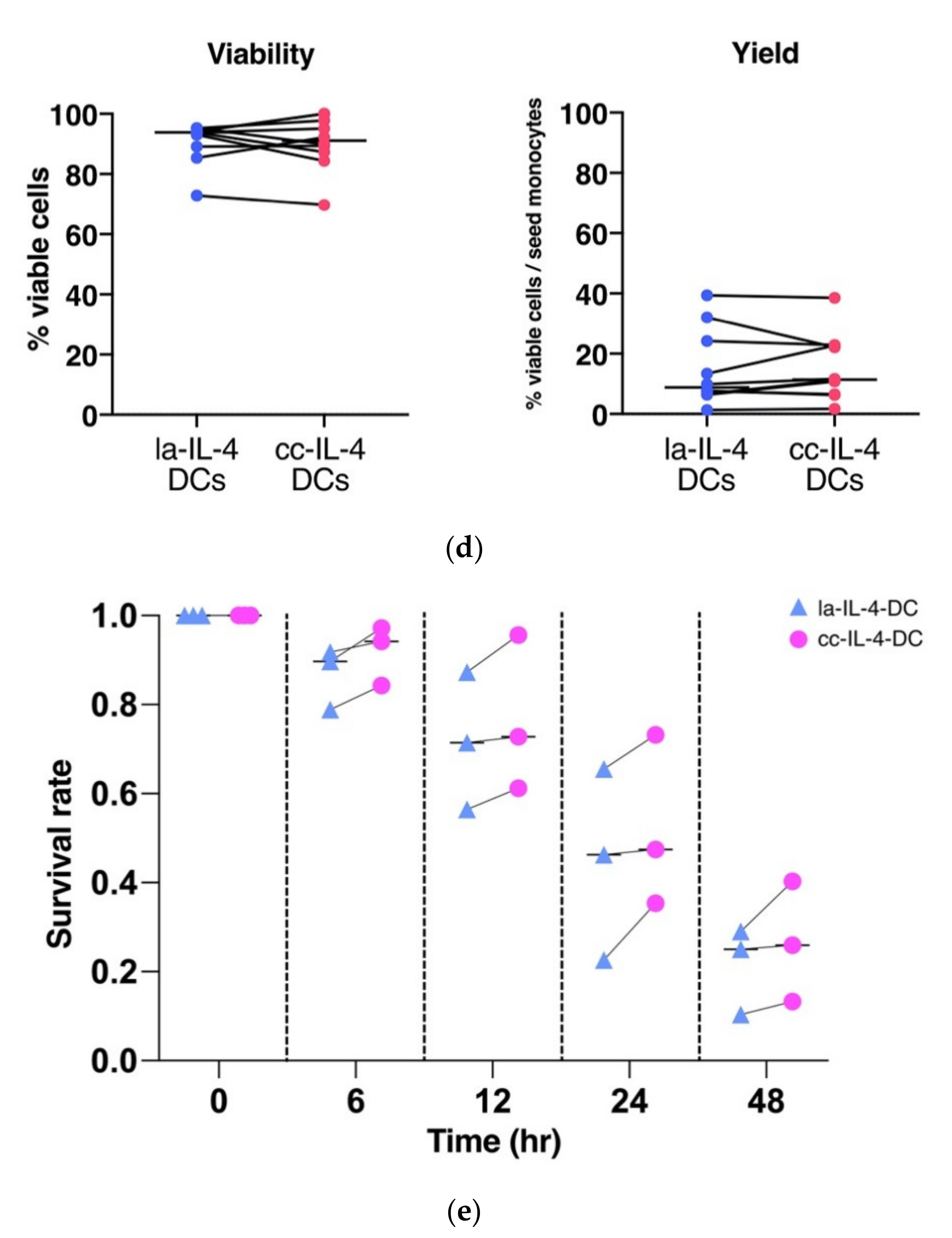
2.4. Cell Survival Analysis
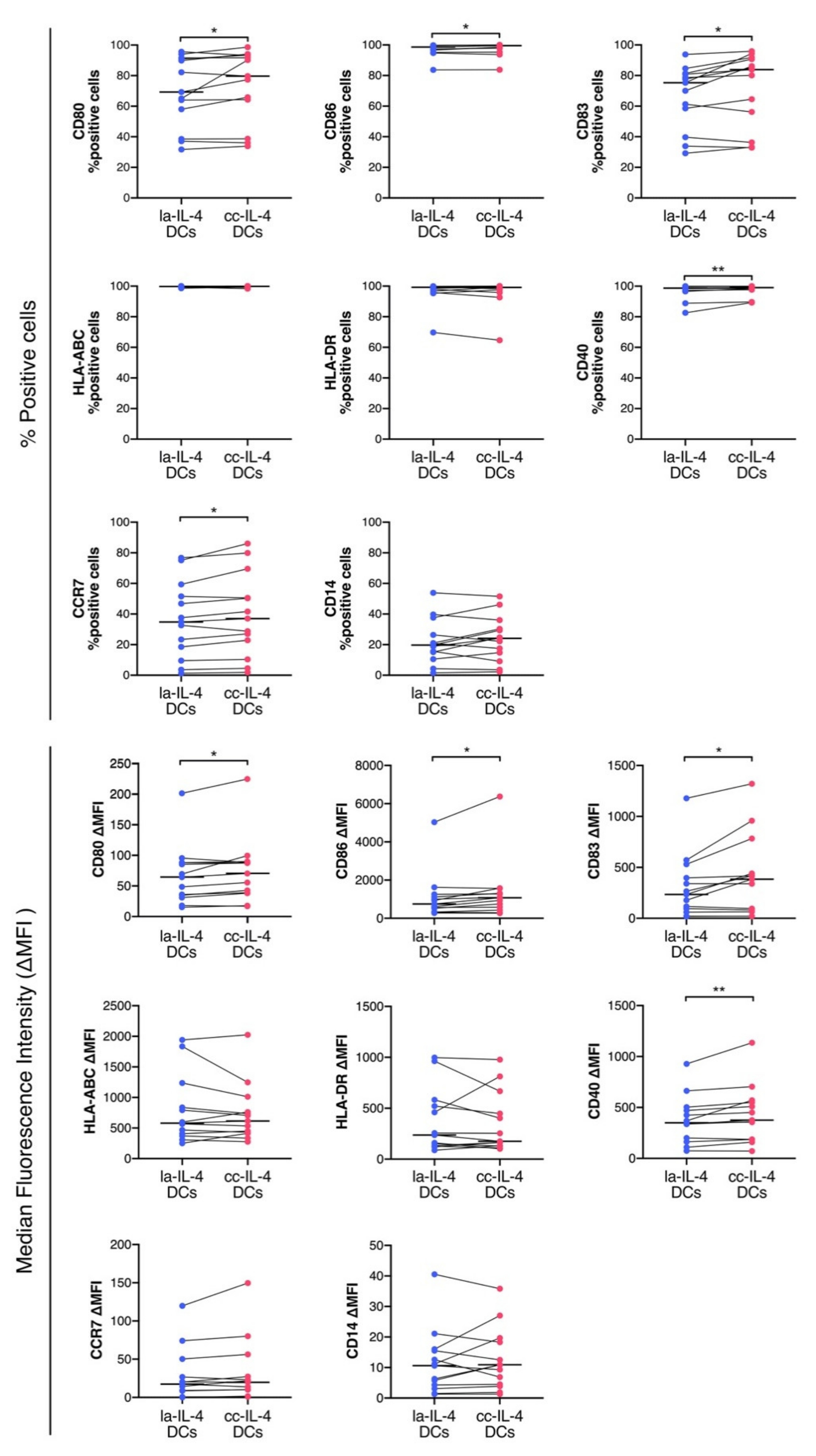
2.5. Surface Marker Analysis of the la−IL-4−DCs and Cluster−Controlled IL-4−DCs
2.6. CTL Induction In Vitro
2.7. Cytokine Production
2.8. RNA Extraction and Microarray Analysis
2.9. Real−Time Reverse Transcription−PCR
2.10. Enzyme−Linked Immunospot (ELISpot) Assays
2.11. Statistical Analysis
3. Results
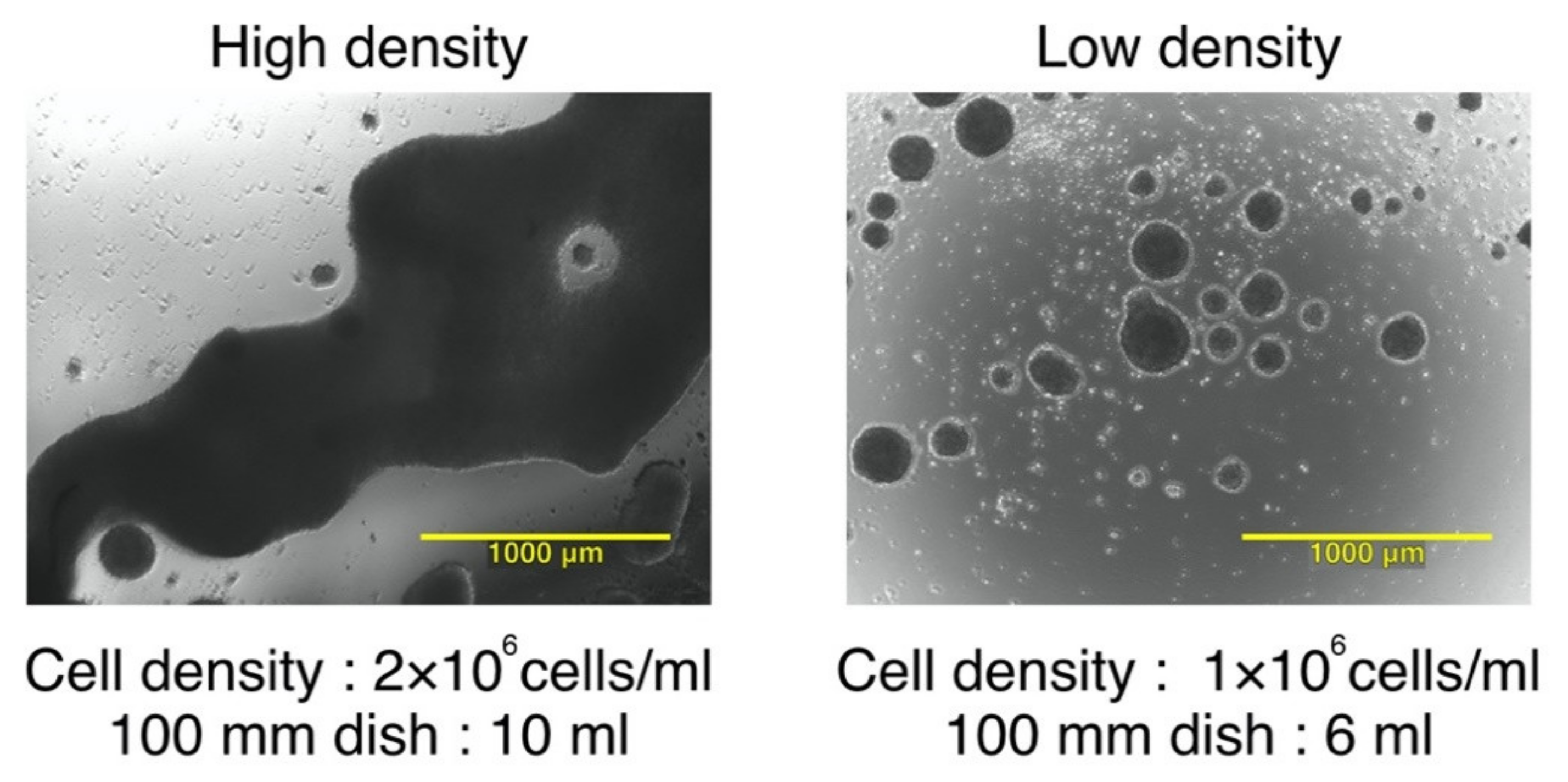
3.1. The Effect of Seeding Density and Total Cell Number on Cluster Formation and Phenotype of IL-4−DCs during Maturation
3.2. Control of Cell Cluster by Culture Dish Increased Expression of Maturation Markers in DCs
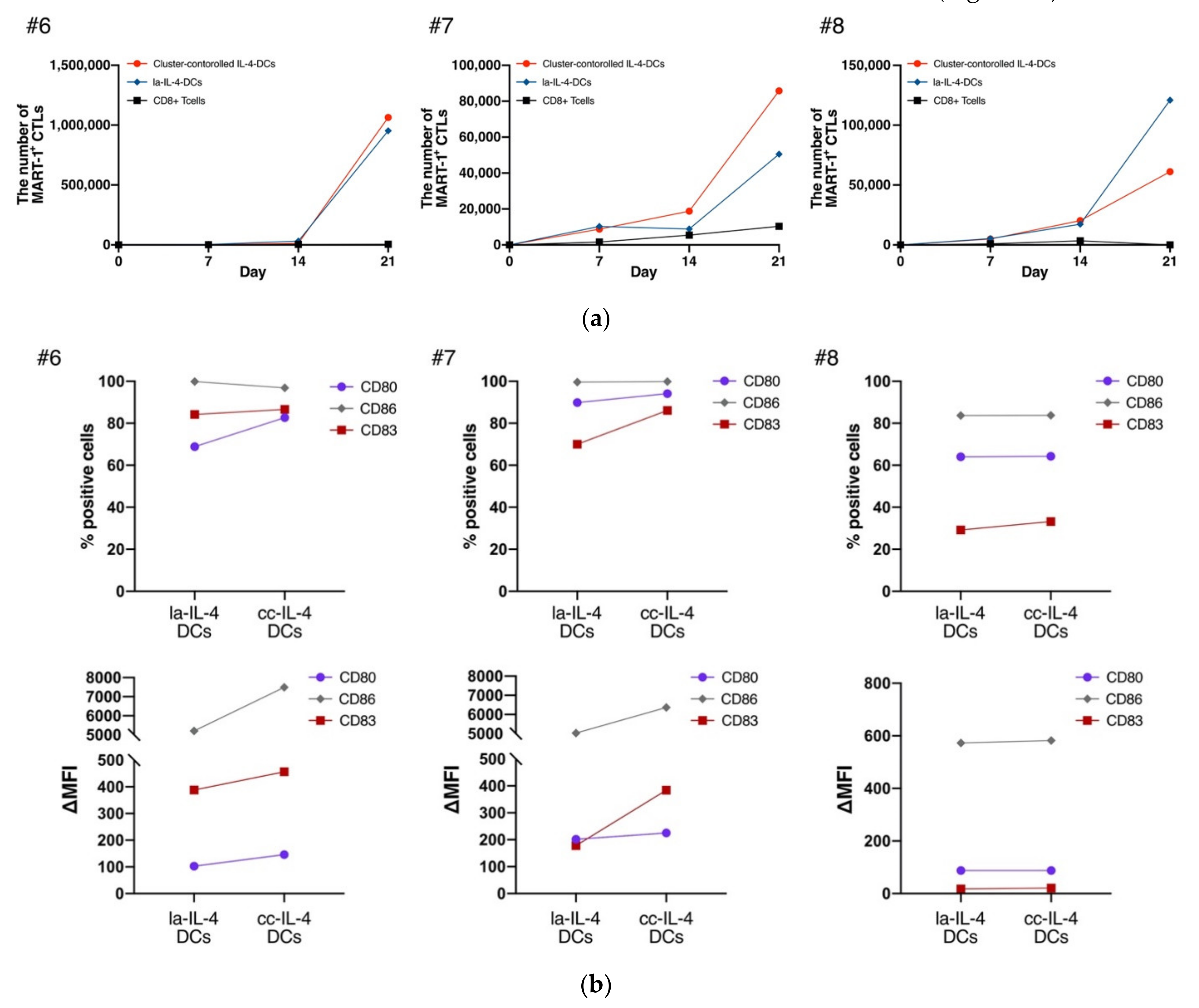
3.3. cc−IL-4−DCs Exhibited the Ability of Presenting Antigens to CD8+ T Cells
3.4. cc−IL-4−DCs Promoted Higher IFN−γ Production Compared with That of la−IL-4−DCs
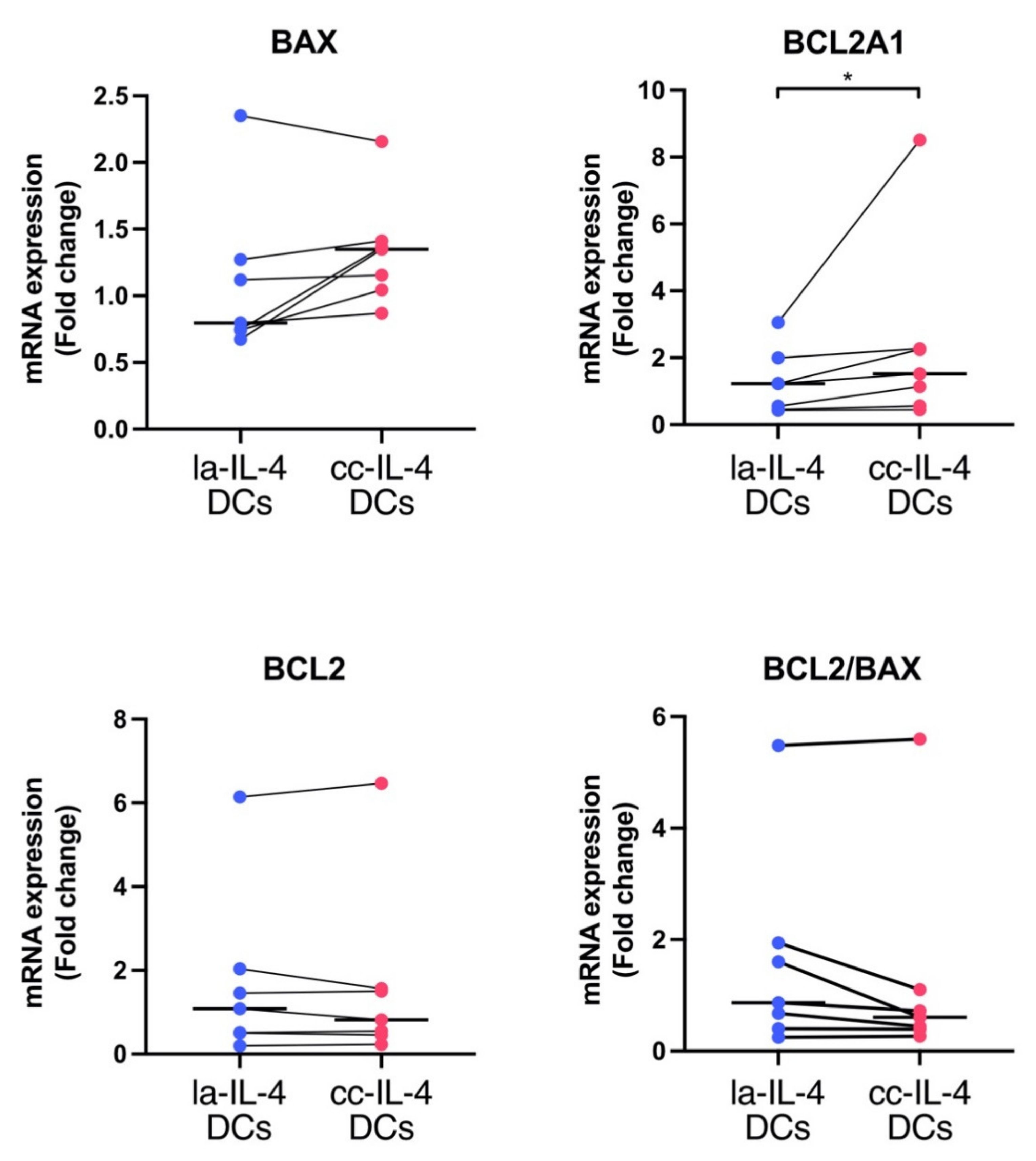
3.5. BCL2A1 Gene Expression in cc−IL-4−DCs Compared with la−IL-4−DCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Zammit, D.J.; Cauley, L.S.; Pham, Q.M.; Lefrançois, L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity 2005, 22, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Smits, E.L.; Lion, E.; Van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, 257–267. [Google Scholar] [CrossRef]
- Figdor, C.G.; De Vries, I.J.M.; Lesterhuis, W.J.; Melief, C.J.M. Dendritic cell immunotherapy: Mapping the way. Nat. Med. 2004, 10, 475–480. [Google Scholar] [CrossRef]
- Santos, P.M.; Butterfield, L.H. Dendritic cell–based cancer vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef]
- Ribas, A.; Comin-Anduix, B.; Chmielowski, B.; Jalil, J.; De La Rocha, P.; McCannel, T.A.; Ochoa, M.T.; Seja, E.; Villanueva, A.; Oseguera, D.K.; et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin. Cancer Res. 2009, 15, 6267–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesselhut, J.; Marx, D.; Lange, H.; Regalo, G.; Cillien, N.; Chang, R.Y.; Nesselhut, T. Systemic treatment with anti-PD-1 antibody nivolumab in combination with vaccine therapy in advanced pancreatic cancer. J. Clin. Oncol. 2016, 34, 3092. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, 5931. [Google Scholar] [CrossRef] [Green Version]
- Draube, A.; Klein-González, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimodaira, S.; Sano, K.; Hirabayashi, K.; Koya, T.; Higuchi, Y.; Mizuno, Y.; Yamaoka, N.; Yuzawa, M.; Kobayashi, T.; Ito, K.; et al. Dendritic cell-based adjuvant vaccination targeting Wilms’ Tumor 1 in patients with advanced colorectal cancer. Vaccines 2015, 3, 1004–1018. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, S.; Yanagisawa, R.; Koya, T.; Hirabayashi, K.; Higuchi, Y.; Sakamoto, T.; Togi, M.; Kato, T.; Kobayashi, T.; Koizumi, T.; et al. In vivo administration of recombinant human granulocyte colony-stimulating factor increases the immune effectiveness of dendritic cell-based cancer vaccination. Vaccines 2019, 7, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroki, H.; Morisaki, T.; Matsumoto, K.; Onishi, H.; Baba, E.; Tanaka, M.; Katano, M. Streptococcal preparation OK-432: A new maturation factor of monocyte-derived dendritic cells for clinical use. Cancer Immunol. Immunother. 2003, 52, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Ryoma, Y.; Moriya, Y.; Okamoto, M.; Kanaya, I.; Saito, M.; Sato, M. Biological effect of OK-432 (Picibanil) and possible application to dendritic cell therapy. Anticancer Res. 2004, 24, 3295–3301. [Google Scholar]
- Pan, K.; Lv, L.; Zheng, H.; Zhao, J.; Pan, Q.; Li, J.; Weng, D.; Wang, D.; Jiang, S.; Chang, A.E.; et al. OK-432 synergizes with IFN-γ to confer dendritic cells with enhanced antitumor immunity. Immunol. Cell Biol. 2014, 92, 263–274. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, L.; Ling, F.; Wen, S.; Luo, Y.; Liu, H.; Liu, J.; Zheng, W.; Liang, M.; Sun, J.; et al. Effect of immune tolerance induced by immature dendritic cells and CTLA4-Ig on systemic lupus erythematosus: An in vivo study. Exp. Ther. Med. 2018, 15, 2499–2506. [Google Scholar] [CrossRef]
- Audiger, C.; Rahman, M.J.; Yun, T.J.; Tarbell, K.V.; Lesage, S. The importance of dendritic cells in maintaining immune tolerance. J. Immunol. 2017, 198, 2223–2231. [Google Scholar] [CrossRef] [Green Version]
- Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. The dendritic cell-regulatory T lymphocyte crosstalk contributes to tumor-induced tolerance. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Date, I.; Koya, T.; Sakamoto, T.; Togi, M.; Kawaguchi, H.; Watanabe, A.; Kato, T.; Shimodaira, S., Jr. Interferon-α-induced dendritic cells generated with human platelet lysate exhibit elevated antigen presenting ability to cytotoxic T lymphocytes. Vaccines 2021, 9, 10. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Palucka, A.K.; Britten, C.M.; Dhodapkar, M.V.; Håkansson, L.; Janetzki, S.; Kawakami, Y.; Kleen, T.O.; Lee, P.P.; Maccalli, C.; et al. Recommendations from the iSBTc-SITC/FDA/NCI workshop on immunotherapy biomarkers. Clin. Cancer Res. 2011, 17, 3064–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimodaira, S.; Terutsugu, K.; Yumiko, H.; Masato, O.; Shigeo, K. Quality verification of dendritic cell-based cancer vaccine. Pharm. Anal. Acta 2015, 7, 8–12. [Google Scholar] [CrossRef]
- Wang, J.; Dai, X.; Hsu, C.; Ming, C.; He, Y.; Zhang, J.; Wei, L.; Zhou, P.; Wang, C.Y.; Yang, J.; et al. Discrimination of the heterogeneity of bone marrow-derived dendritic cells. Mol. Med. Rep. 2017, 16, 6787–6793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koya, T.; Date, I.; Kawaguchi, H.; Watanabe, A.; Sakamoto, T.; Togi, M.; Kato, T.; Yoshida, K.; Kojima, S.; Yanagisawa, R.; et al. Dendritic cells pre-pulsed with Wilms’ Tumor 1 in optimized culture for cancer vaccination. Pharmaceutics 2020, 12, 305. [Google Scholar] [CrossRef] [Green Version]
- Sauter, A.; Mc Duffie, Y.; Boehm, H.; Martinez, A.; Spatz, J.P.; Appel, S. Surface-mediated priming during in vitro generation of monocyte-derived dendritic cells. Scand. J. Immunol. 2015, 81, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Sauter, A.; Yi, D.H.; Li, Y.; Roersma, S.; Appel, S. The culture dish surface influences the phenotype and cytokine production of human monocyte-derived dendritic cells. Front. Immunol. 2019, 10, 2352. [Google Scholar] [CrossRef] [PubMed]
- Cesarz, Z.; Tamama, K. Spheroid culture of mesenchymal stem cells. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Ryu, N.-E.; Lee, S.-H.; Park, H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, C.; Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Mueller-Klieser, W. Three-dimensional cell cultures: From molecular mechanisms to clinical applications. Am. J. Physiol. Physiol. 1997, 273. [Google Scholar] [CrossRef]
- Sasai, Y. Next-generation regenerative medicine: Organogenesis from stem cells in 3D culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, J.E.; Thomson, B.; Genever, P.G. Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng. Part C Methods 2010, 16, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Bhang, S.H.; Lee, S.; Shin, J.Y.; Lee, T.J.; Kim, B.S. Transplantation of cord blood mesenchymal stem cells as spheroids enhances vascularization. Tissue Eng. Part A 2012, 18, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.C.; Liu, Y.; Yuan, X.; Ma, T. Compaction, fusion, and functional activation of three-dimensional human mesenchymal stem cell aggregate. Tissue Eng. Part A 2015, 21, 1705–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettinato, G.; Ramanathan, R.; Fisher, R.A.; Mangino, M.J.; Zhang, N.; Wen, X. Scalable differentiation of human iPSCs in a multicellular spheroid-based 3D culture into hepatocyte-like cells through direct Wnt/β-catenin pathway inhibition. Sci. Rep. 2016, 6, 32888. [Google Scholar] [CrossRef]
- Torizal, F.G.; Kimura, K.; Horiguchi, I.; Sakai, Y. Size-dependent hepatic differentiation of human induced pluripotent stem cells spheroid in suspension culture. Regen. Ther. 2019, 12, 66–73. [Google Scholar] [CrossRef]
- Shimodaira, S.; Hirabayashi, K.; Kobayashi, T.; Higuchi, Y.; Yokokawa, K. Future prospective of cancer vaccination technology in Japan. Pharm. Reg. Aff. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Higuchi, Y.; Koya, T.; Yuzawa, M.; Yamaoka, N.; Mizuno, Y.; Yoshizawa, K.; Hirabayashi, K.; Kobayashi, T.; Sano, K.; Shimodaira, S. Enzyme-linked immunosorbent spot assay for the detection of wilms’ tumor 1-specific T cells induced by dendritic cell vaccination. Biomedicines 2015, 3, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Nagayama, H.; Tadokoro, K.; Juji, T.; Takahashi, T.A. Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38 mapk are involved in IL-10-mediated selective repression of TNF-alpha-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J. Immunol. 1999, 162, 3865–3872. [Google Scholar] [PubMed]
- Schmidt, S.V.; Nino-Castro, A.C.; Schultze, J.L. Regulatory dendritic cells: There is more than just immune activation. Front. Immunol. 2012, 3, 274. [Google Scholar] [CrossRef] [Green Version]
- Aerts-Toegaert, C.; Heirman, C.; Tuyaerts, S.; Corthals, J.; Aerts, J.L.; Bonehill, A.; Thielemans, K.; Breckpot, K.; Breckpot, K. CD83 expression on dendritic cells and T cells: Correlation with effective immune responses. Eur. J. Immunol. 2007, 37, 686–695. [Google Scholar] [CrossRef]
- Hill, K.S.; Errington, F.; Steele, L.P.; Merrick, A.; Morgan, R.; Selby, P.J.; Georgopoulos, N.T.; O’Donnell, D.M.; Melcher, A.A. OK432-activated human dendritic cells kill tumor cells via CD40/CD40 ligand interactions. J. Immunol. 2008, 181, 3108–3115. [Google Scholar] [CrossRef]
- Nakahara, S.; Tsunoda, T.; Baba, T.; Asabe, S.; Tahara, H. Dendritic cells stimulated with a bacterial product, OK-432, efficiently induce cytotoxic T lymphocytes specific to tumor rejection peptide. Cancer Res. 2003, 63, 4112–4118. [Google Scholar]
- Sato, M.; Takayama, T.; Tanaka, H.; Konishi, J.; Suzuki, T.; Kaiga, T.; Tahara, H. Generation of mature dendritic cells fully capable of T helper type 1 polarization OK-432 combined with prostaglandin E2. Cancer Sci. 2003, 94, 1091–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.C.; Whitehead, J.; Falahee, P.C.; Zhou, D.; Simon, S.I.; Leach, J.K. Multifactorial experimental design to optimize the anti-inflammatory and proangiogenic potential of mesenchymal stem cell spheroids. Stem Cells 2017, 35, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Ouaaz, F.; Arron, J.; Zheng, Y.; Choi, Y.; Beg, A.A. Dendritic cell development and survival require distinct NF-κB subunits. Immunity 2002, 16, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Olsson Åkefeldt, S.; Maisse, C.; Belot, A.; Mazzorana, M.; Salvatore, G.; Bissay, N.; Jurdic, P.; Aricò, M.; Rabourdin-Combe, C.; Henter, J.-I.; et al. Chemoresistance of human monocyte-derived dendritic cells is regulated by IL-17A. PLoS ONE 2013, 8, e56865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, C.; Zhu, F.; Liu, F.; Zhang, P.; Guo, C.; Wang, X.; Li, H.; Ma, C.; Sun, W.; et al. Reoxygenation of hypoxia-differentiated dentritic cells induces Th1 and Th17 cell differentiation. Mol. Immunol. 2010, 47, 922–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Markers | Median of % Positive Cells (Minimum–Maximum) | Median Fluorescence Intensity (ΔMFI) (Minimum–Maximum) | ||
|---|---|---|---|---|
| High Density | Low Density | High Density | Low Density | |
| CD80 | 68.9 | 73.5 | 11 | 17.2 |
| (41.6–78.5) | (39.2–91.8) | (4.0–26.6) | (2.4–24.5) | |
| CD86 | 97.1 | 97.2 | 151.6 | 297.1 |
| (92.8–98.1) | (91.3–97.6) | (75.2–367.0) | (56.6–305.9) | |
| CD83 | 55.3 | 68.6 | 7 | 11.5 |
| (53.4–60.9) | (46.5–69.9) | (7.0–14.1) | (5.3–17.1) | |
| CD40 | 98.2 | 96.8 | 66 | 95.7 |
| (94.9–98.5) | (93.1–99.3) | (43.0–151.8) | (42.1–153.1) | |
| CCR7 | 33.7 | 36.7 | 2.7 | 3.3 |
| (26.1–38.8) | (22.9–62.0) | (2.0–5.6) | (1.8–6.8) | |
| HLA−ABC | 99.2 | 99.4 | 137.6 | 179.7 |
| (96.2–99.7) | (98.6–99.8) | (52.7–174.8) | (149.1–206.6) | |
| HLA−DR | 99.3 | 99.8 | 361 | 519.6 |
| (89.0–99.8) | (99.1–99.9) | (54.9–798.3) | (99.1–1059.0) | |
| CD11c | 99.8 | 99.5 | 204.9 | 193.6 |
| (99.6–99.9) | (99.4–99.8) | (136.0–312.0) | (161.4–282.0) | |
| CD14 | 29.2 | 20.9 | 3.2 | 1.8 |
| (8.1–44.3) | (13.0–27.2) | (1.0–5.1) | (1.2–3.6) | |
| Gene Symbol | Patient #1 | Patient #2 | Patient #3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DC Preparation | Fold Change vs. Controls | DC Preparation | Fold Change vs. Controls | DC Preparation | Fold Changevs. Controls | ||||
| la−IL-4−DCs | cc−IL-4−DCs | la−IL-4−DCs | cc−IL-4−DCs | la−IL-4−DCs | cc−IL-4−DCs | ||||
| BCL2 | 138.5 | 162.9 | 1.2 | 82.1 | 81.6 | 1.0 | 158.5 | 141.9 | 0.9 |
| BCL2L1 | 94.8 | 97.9 | 1.0 | 80.2 | 80.4 | 1.0 | 53.3 | 50.0 | 0.9 |
| BCL2L2 | 91.6 | 74.7 | 0.9 | 70.2 | 77.2 | 1.1 | 73.2 | 50.1 | 0.7 |
| MCL1 | 610.8 | 686.3 | 1.1 | 879.6 | 772.1 | 0.9 | 906.7 | 913.8 | 1.0 |
| BCL2A1 | 695.0 | 977.8 | 1.4 | 486.9 | 650.4 | 1.3 | 233.3 | 447.3 | 1.9 |
| BAX | 99.9 | 100.1 | 1.0 | 94.6 | 95.8 | 1.0 | 87.7 | 81.6 | 0.9 |
| BOK | 24.4 | 27.4 | 1.1 | 24.9 | 25.0 | 1.0 | 27.2 | 24.9 | 0.9 |
| BAK1 | 125.3 | 124.2 | 1.0 | 124.0 | 138.8 | 1.1 | 134.6 | 135.3 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawaguchi, H.; Sakamoto, T.; Koya, T.; Togi, M.; Date, I.; Watanabe, A.; Yoshida, K.; Kato, T.; Nakamura, Y.; Ishigaki, Y.; et al. Quality Verification with a Cluster−Controlled Manufacturing System to Generate Monocyte−Derived Dendritic Cells. Vaccines 2021, 9, 533. https://doi.org/10.3390/vaccines9050533
Kawaguchi H, Sakamoto T, Koya T, Togi M, Date I, Watanabe A, Yoshida K, Kato T, Nakamura Y, Ishigaki Y, et al. Quality Verification with a Cluster−Controlled Manufacturing System to Generate Monocyte−Derived Dendritic Cells. Vaccines. 2021; 9(5):533. https://doi.org/10.3390/vaccines9050533
Chicago/Turabian StyleKawaguchi, Haruhiko, Takuya Sakamoto, Terutsugu Koya, Misa Togi, Ippei Date, Asuka Watanabe, Kenichi Yoshida, Tomohisa Kato, Yuka Nakamura, Yasuhito Ishigaki, and et al. 2021. "Quality Verification with a Cluster−Controlled Manufacturing System to Generate Monocyte−Derived Dendritic Cells" Vaccines 9, no. 5: 533. https://doi.org/10.3390/vaccines9050533
APA StyleKawaguchi, H., Sakamoto, T., Koya, T., Togi, M., Date, I., Watanabe, A., Yoshida, K., Kato, T., Nakamura, Y., Ishigaki, Y., & Shimodaira, S. (2021). Quality Verification with a Cluster−Controlled Manufacturing System to Generate Monocyte−Derived Dendritic Cells. Vaccines, 9(5), 533. https://doi.org/10.3390/vaccines9050533