
Acute Kidney Injury in Monoclonal Gammopathies

 , and
, and
Abstract

1. Introduction
2. Acute Kidney Injury in MG: Pathogenesis and Clinical Presentation
3. Acute Kidney Injury: Risk Factors and Their Relationship to MG
4. Handling of MC Proteins by the Kidney—Tubular Toxicity of Filtered Proteins
5. Tubular Obstructive Nephropathy
6. Crystal Precipitation of MC Proteins
7. The Effects of Hypercalcemia on Kidney Function
8. AKI Related to Chemotherapy of MGRS/Multiple Myeloma
9. The Burden of Age: Epidemiology of AKI Related to MG
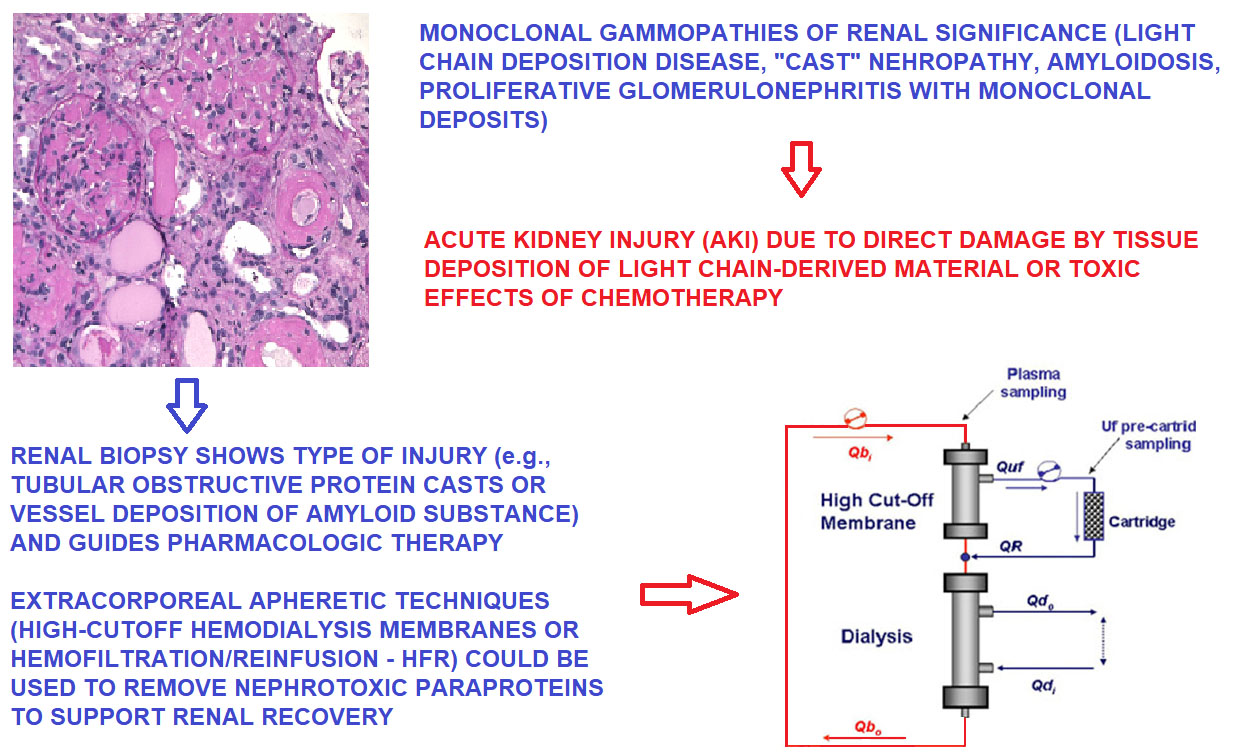
10. Renal Biopsy: Linking Persistent Urinary Abnormalities and AKI or Progressive Renal Failure to MGRS
11. Treatment of AKI by Renal Apheresis: High-Cutoff Membrane Hemodialysis
12. Treatment of AKI by Renal Apheresis: Hemofiltration with Endogenous Reinfusion
13. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef]
- Mouhieddine, T.H.; Weeks, L.; Ghobrial, I.M. Monoclonal gammopathy of undetermined significance. Blood 2019, 133, 2484–2494. [Google Scholar] [CrossRef]
- Atkin, C.; Reddy-Kolanu, V.; Drayson, M.T.; Sapey, E.; Richter, A.G. The prevalence and significance of monoclonal gammopathy of undetermined significance in acute medical admissions. Br. J. Haematol. 2020, 189, 1127–1135. [Google Scholar] [CrossRef]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and prevalence of monoclonal gammopathy of undetermined significance: A study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Leung, N.; Bridoux, F.; Hutchison, C.A.; Nasr, S.H.; Cockwell, P.; Fermand, J.-P.; Dispenzieri, A.; Song, K.W.; Kyle, R.A. Monoclonal gammopathy of renal significance: When MGUS is no longer undetermined or insignificant. Blood 2012, 120, 4292–4295. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Motwani, S.S.; Herlitz, L.; Monga, D.; Jhaveri, K.D.; Lam, A.Q. Paraprotein-related kidney disease: Glomerular diseases asso-ciated with paraproteinemias. Clin. J. Am. Soc. Nephrol. 2016, 11, 2260. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Rajkumar, S.V.; D’Agati, V.D. The complexity and heterogeneity of monoclonal immunoglobulin–associated renal diseases. J. Am. Soc. Nephrol. 2018, 29, 1810–1823. [Google Scholar] [CrossRef]
- Doshi, M.; Lahoti, A.; Danesh, F.; Batuman, V.; Sanders, P.W. Paraprotein–related kidney disease: Kidney injury from paraproteins—what determines the site of injury? Clin. J. Am. Soc. Nephrol. 2016, 11, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Bridoux, F.; Nasr, S.H. Monoclonal gammopathy of renal significance. N. Engl. J. Med. 2021, 384, 1931–1941. [Google Scholar] [CrossRef]
- Menè, P.; De Alexandris, L.; Moioli, A.; Raffa, S.; Stoppacciaro, A. Monoclonal gammopathies of renal significance: Renal biopsy and beyond. Cancers 2020, 12, 1741. [Google Scholar] [CrossRef]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Prim. 2018, 4, 38. [Google Scholar] [CrossRef]
- Rosner, M.H.; Perazella, M.A. Acute kidney injury in the patient with cancer. Kidney Res. Clin. Pract. 2019, 38, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Asonitis, N.; Angelousi, A.; Zafeiris, C.; I Lambrou, G.; Dontas, I.; Kassi, E. Diagnosis, Pathophysiology and Management of Hypercalcemia in Malignancy: A Review of the Literature. Horm. Metab. Res. 2019, 51, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Mirrakhimov, A.E. Hypercalcemia of malignancy: An update on pathogenesis and management. North Am. J. Med. Sci. 2015, 7, 483–493. [Google Scholar] [CrossRef]
- Leung, N.; Drosou, M.E.; Nasr, S.H. Dysproteinemias and glomerular disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Herlitz, L.C.; D’Agati, V.D.; Markowitz, G.S. Crystalline nephropathies. Arch. Pathol. Lab. Med. 2012, 136, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Wanchoo, R.; Abudayyeh, A.; Doshi, M.; Edeani, A.; Glezerman, I.G.; Monga, D.; Rosner, M.; Jhaveri, K.D. Renal toxicities of novel agents used for treatment of multiple myeloma. Clin. J. Am. Soc. Nephrol. 2017, 12, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Lameire, N.; KDIGO AKI Guideline Work Group. Diagnosis, evaluation, and management of acute kidney injury: A KDIGO summary (Part 1). Crit. Care 2013, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, R.; Sciascia, S.; Baldovino, S.; Roccatello, D. Acute kidney injury associated with glomerular diseases. Curr. Opin. Crit. Care 2019, 25, 573. [Google Scholar] [CrossRef]
- Heaney, J.L.J.; Campbell, J.P.; Yadav, P.; Griffin, A.E.; Shemar, M.; Pinney, J.H.; Drayson, M.T. Multiple myeloma can be accurately diagnosed in acute kidney injury patients using a rapid serum free light chain test. BMC Nephrol. 2017, 18, 247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Royal, V.; Leung, N.; Troyanov, S.; Nasr, S.H.; Écotière, L.; Leblanc, R.; Adam, B.; Angioi, A.; Alexander, M.P.; Asunis, A.M.; et al. Clinicopathologic predictors of renal outcomes in light chain cast nephropathy: A multicenter retrospective study. Blood 2020, 135, 1833–1846. [Google Scholar] [CrossRef]
- Yadav, P.; Sathick, I.J.; Leung, N.; Brown, E.E.; Cook, M.; Sanders, P.; Cockwell, P. Serum free light chain level at diagnosis in myeloma cast nephropathy—A multicentre study. Blood Cancer J. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Picken, M.M. Amyloidosis-where are we now and where are we heading? Arch. Pathol. Lab. Med. 2010, 134, 545–551. [Google Scholar] [CrossRef]
- Picken, M.M.; Herrera, G.A.; Dogan, A. (Eds.) Amyloid and Related Disorders; Humana Press: Totowa, NJ, USA, 2015. [Google Scholar]
- Vaxman, I.; Gertz, M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020, 143, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Thajudeen, B.; Rubin, M.F. Hemodynamic acute kidney injury in immunoglobulin A nephropathy: Nephrosarca theory revisited. Am. J. Med. 2013, 126, e13–e14. [Google Scholar] [CrossRef]
- Oyama, Y.; Iwafuchi, Y.; Morioka, T.; Narita, I. Acute kidney injury associated with minimal change nephrotic syndrome in an elderly patient successfully treated with both fluid management and specific therapy based on kidney biopsy findings. Case Rep. Nephrol. Dial. 2020, 10, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yin, Y.; Jin, L.; Zhang, R.; Hou, Z.; Zhang, Y. Acute compartment syndrome. Medicine (Baltimore) 2019, 98, e16260. [Google Scholar] [CrossRef]
- Khanna, A. Acquired nephrogenic diabetes insipidus. Semin. Nephrol. 2006, 26, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Khositseth, S.; Charngkaew, K.; Boonkrai, C.; Somparn, P.; Uawithya, P.; Chomanee, N.; Payne, D.M.; Fenton, R.; Pisitkun, T. Hypercalcemia induces targeted autophagic degradation of aquaporin-2 at the onset of nephrogenic diabetes insipidus. Kidney Int. 2017, 91, 1070–1087. [Google Scholar] [CrossRef]
- Asghar, M.; Ahmed, K.; Shah, S.; Siddique, M.; Dasgupta, P.; Khan, M. Renal vein thrombosis. Eur. J. Vasc. Endovasc. Surg. 2007, 34, 217–223. [Google Scholar] [CrossRef]
- Mirrakhimov, A.E.; Ali, A.M.; Barbaryan, A.; Prueksaritanond, S.; Hussain, N. Primary nephrotic syndrome in adults as a risk factor for pulmonary embolism: An up-to-date review of the literature. Int. J. Nephrol. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Magni, A.; Agostoni, P.; Bonezzi, C.; Massazza, G.; Menè, P.; Savarino, V.; Fornasari, D. Management of osteoarthritis: Expert opinion on NSAIDs. Pain Ther. 2021, 1–26. [Google Scholar] [CrossRef]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of origin and genetic alterations in the pathogenesis of multiple mye-loma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [PubMed]
- Nezlin, R. Dynamic aspects of the immunoglobulin structure. Immunol. Investig. 2019, 48, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Birn, H.; Storm, T.; Weyer, K.; Nielsen, R. Endocytic receptors in the renal proximal tubule. Physiology (Be-thesda) 2012, 27, 223. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Eshbach, M.L.; Weisz, O.A. Receptor-mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef]
- Dickson, L.E.; Wagner, M.C.; Sandoval, R.M.; Molitoris, B.A. The proximal tubule and albuminuria: Really! J. Am. Soc. Nephrol. 2014, 25, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Stringer, S.; Basnayake, K.; Hutchison, C.; Cockwell, P. Recent advances in the pathogenesis and management of cast nephropathy (myeloma kidney). Bone Marrow Res. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leboulleux, M.; Lelongt, B.; Mougenot, B.; Touchard, G.; Makdassi, R.; Rocca, A.; Noel, L.-H.; Ronco, P.M.; Aucouturier, P. Protease resistance and binding of Ig light chains in myeloma-associated tubulopathies. Kidney Int. 1995, 48, 72–79. [Google Scholar] [CrossRef]
- Yu, X.-J.; Zhou, X.-J.; Wang, S.-X.; Zhou, F.-D.; Zhao, M.-H. Monoclonal light chain crystalline podocytopathy and tubulopathy associated with monoclonal gammopathy of renal significance: A case report and literature review. BMC Nephrol. 2018, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; El Ters, M.; Kashani, K.; Leung, N.; Nasr, S.H. Crystalglobulin-induced nephropathy. J. Am. Soc. Nephrol. 2015, 26, 525–529. [Google Scholar] [CrossRef]
- Rahmani, B.; Patel, S.; Seyam, O.; Gandhi, J.; Reid, I.; Smith, N.; Khan, S.A. Current understanding of tumor lysis syndrome. Hematol. Oncol. 2019, 37, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Batuman, V. Proximal tubular injury in myeloma. Contrib. Nephrol. 2007, 153, 87–104. [Google Scholar] [CrossRef]
- Herrera, G.A. Proximal tubulopathies associated with monoclonal light chains: The spectrum of clinicopathologic manifestations and molecular pathogenesis. Arch. Pathol. Lab. Med. 2014, 138, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.B.; Valeri, A.M.; Herlitz, L.C.; Khan, A.M.; Siegel, D.S.; Markowitz, G.S.; D’Agati, V.D. Light chain proximal tubulopathy: Clinical and pathologic characteristics in the modern treatment era. J. Am. Soc. Nephrol. 2015, 27, 1555–1565. [Google Scholar] [CrossRef]
- Messiaen, T.; Deret, S.; Mougenot, B.; Bridoux, F.; Dequiedt, P.; Dion, J.-J.; Makdassi, R.; Meeus, F.; Pourrat, J.; Touchard, G.; et al. Adult faconi syndrome secondary to light chain gammopathy: Clinicopathologic Heterogeneity and unnsual features in 11 patients. Medicine 2000, 79, 135–154. [Google Scholar] [CrossRef]
- Sanders, P.; Booker, B.B.; Bishop, J.B.; Cheung, H.C. Mechanisms of intranephronal proteinaceous cast formation by low molecular weight proteins. J. Clin. Investig. 1990, 85, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Booker, B.B. Pathobiology of cast nephropathy from human Bence Jones proteins. J. Clin. Investig. 1992, 89, 630–639. [Google Scholar] [CrossRef]
- Glezerman, I.G.; Sternlicht, H. Hypercalcemia of malignancy and new treatment options. Ther. Clin. Risk Manag. 2015, 11, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Brunello, A.; Roma, A.; Basso, U. Medical treatment of malignancy-associated hypercalcemia. Curr. Med. Chem. 2008, 15, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Abudayyed, A. Onconephrology: An evolving field. Methodist DeBakey Cardiovasc. J. 2019, 15, 305–307. [Google Scholar] [CrossRef]
- Stewart, A.F. Hypercalcemia associated with cancer. N. Engl. J. Med. 2005, 352, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.J. Hypercalcaemia—presentation and management. Clin. Med. 2017, 17, 270–273. [Google Scholar] [CrossRef]
- Sargent, J.T.S.; Smith, O.P. Haematological emergencies managing hypercalcaemia in adults and children with haematological disorders. Br. J. Haematol. 2010, 149, 465–477. [Google Scholar] [CrossRef]
- Chang, W.-T.W.; Radin, B.; McCurdy, M.T. Calcium, magnesium, and phosphate abnormalities in the emergency department. Emerg. Med. Clin. North Am. 2014, 32, 349–366. [Google Scholar] [CrossRef]
- Goldner, W. Cancer-related hypercalcemia. J. Oncol. Pract. 2016, 12, 426–432. [Google Scholar] [CrossRef]
- Zagzag, J.; Hu, M.I.; Fisher, S.B.; Perrier, N.D. Hypercalcemia and cancer: Differential diagnosis and treatment. CA Cancer J. Clin. 2018, 68, 377. [Google Scholar] [CrossRef] [PubMed]
- Legrand, S.B.; Leskuski, D.; Zama, I. Narrative review: Furosemide for hypercalcemia: An unproven yet common practice. Ann. Intern. Med. 2008, 149, 259–263. [Google Scholar] [CrossRef]
- Cicci, J.D.; Buie, L.; Bates, J.; Van Deventer, H. Denosumab for the management of hypercalcemia of malignancy in patients with multiple myeloma and renal dysfunction. Clin. Lymphoma Myeloma Leuk. 2014, 14, e207–e211. [Google Scholar] [CrossRef]
- Hu, M.I.; Glezerman, I.; Leboulleux, S.; Insogna, K.; Gucalp, R.; Misiorowski, W.; Yu, B.; Zorsky, P.; Tosi, D.; Bessudo, A.; et al. Denosumab for treatment of hypercalcemia of malignancy. J. Clin. Endocrinol. Metab. 2014, 99, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Dalkin, A.C. Onco-Nephrology: The pathophysiology and treatment of malignancy-associated hypercalcemia. Clin. J. Am. Soc. Nephrol. 2012, 7, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A.; Moeckel, G. Nephrotoxicity from chemotherapeutic agents: Clinical manifestations, pathobiology, and prevention/therapy. Semin. Nephrol. 2010, 30, 570–581. [Google Scholar] [CrossRef]
- Milani, P.; Merlini, G.; Palladini, G. Novel therapies in light chain amyloidosis. Kidney Int. Rep. 2018, 3, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.-P.; Gibbs, S.; et al. The evaluation of monoclonal gammopathy of renal significance: A consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 45–59. [Google Scholar] [CrossRef]
- Rossi, A.C. Emerging options for combination therapy in multiple myeloma. Clin. Adv. Hematol. Oncol. 2018, 16, 192–194. [Google Scholar]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef]
- Kazandjian, D.; Landgren, O. Delaying the use of high-dose melphalan with stem cell rescue in multiple myeloma is ready for prime time. Clin. Adv. Hematol. Oncol. 2019, 17, 559–568. [Google Scholar]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed]
- Fraiser, L.H.; Kanekal, S.; Kehrer, J.P. Cyclophosphamide toxicity. Drugs 1991, 42, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.; Goldstein, J.; Schiff, J.; John, R. Collapsing glomerulopathy following anthracycline therapy. Am. J. Kidney Dis. 2013, 61, 778–781. [Google Scholar] [CrossRef]
- Cosmai, L.; Porta, C.; Gallieni, M.; Perazella, M.A. Onco-nephrology: A decalogue. Nephrol. Dial. Transplant. 2015, 31, 515–519. [Google Scholar] [CrossRef]
- Herrera, G.A.; Joseph, L.; Gu, X.; Hough, A.; Barlogie, B. Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch. Pathol. Lab. Med. 2004, 128, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Joh, K. Pathology of glomerular deposition diseases. Pathol. Int. 2007, 57, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Herrera, G.A.; Picken, M.M. Renal diseases associated with plasma cell dyscrasias, amyloidoses, and Waldenström macro-globulinemia. In Heptinstall’s Pathology of the Kidney, 7th ed.; Jennette, J.C., Olson, J.L., Silva, F.G., D’Agati, V.D., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2014; Chapter 22; pp. 951–1014. [Google Scholar]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368. [Google Scholar] [CrossRef]
- Ramirez-Alvarado, M.; Ward, C.J.; Huang, B.Q.; Gong, X.; Hogan, M.C.; Madden, B.J.; Charlesworth, M.C.; Leung, N. Differences in immunoglobulin light chain species found in urinary exosomes in light chain amyloidosis (AL). PLoS ONE 2012, 7, e38061. [Google Scholar] [CrossRef]
- Ramirez-Alvarado, M.; Barnidge, D.R.; Murray, D.L.; Dispenzieri, A.; Marin-Argany, M.; Dick, C.J.; Cooper, S.A.; Nasr, S.H.; Ward, C.J.; Dasari, S.; et al. Assessment of renal response with urinary exosomes in patients with AL amyloidosis: A proof of concept. Am. J. Hematol. 2017, 92, 536–541. [Google Scholar] [CrossRef]
- Picken, M.M. Proteomics and mass spectrometry in the diagnosis of renal amyloidosis. Clin. Kidney J. 2015, 8, 665–672. [Google Scholar] [CrossRef]
- Herrera, G.A. The value of ultrastructural evaluation in medical renal diseases. Ultrastruct. Pathol. 2019, 43, 225–228. [Google Scholar] [CrossRef]
- Rosenstock, J.L.; Markowitz, G.S.; Valeri, A.M.; Sacchi, G.; Appel, G.B.; D’Agati, V.D. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003, 63, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Satoskar, A.; Markowitz, G.S.; Valeri, A.M.; Appel, G.B.; Stokes, M.; Nadasdy, T.; D’Agati, V.D. Proliferative glomerulonephritis with monoclonal IgG deposits. J. Am. Soc. Nephrol. 2009, 20, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Guiard, E.; Karras, A.; Plaisier, E.; Van Huyen, J.-P.D.; Fakhouri, F.; Rougier, J.-P.; Noel, L.-H.; Callard, P.; Delahousse, M.; Ronco, P.; et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: Correlation with IgG subclass and response to rituximab. Clin. J. Am. Soc. Nephrol. 2011, 6, 1609–1616. [Google Scholar] [CrossRef]
- Roccatello, D.; Saadoun, D.; Ramos-Casals, M.; Tzioufas, A.G.; Fervenza, F.C.; Cacoub, P.; Zignego, A.L.; Ferri, C. Cryoglobulinaemia. Nat. Rev. Dis. Prim. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Frémeaux-Bacchi, V.; Lequintrec, M.; Salomon, R.; Blouin, J.; Knebelmann, B.; Grünfeld, J.P.; Lesavre, P.; Noël, L.H.; Fakhouri, F. Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J. Med. Genet. 2007, 44, 193. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Messias, N.C.; Walker, P.D.; Fidler, M.E.; Cornell, L.D.; Hernandez, L.H.; Alexander, M.P.; Sethi, S.; Nasr, S.H. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. 2015, 88, 867–873. [Google Scholar] [CrossRef]
- Pickering, M.C.; D’Agati, V.D.; Nester, C.M.; Smith, R.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef]
- Pirozzi, N.; Stoppacciaro, A.; Menè, P. Dominant C3 glomerulopathy: New roles for an old actor in renal pathology. J. Nephrol. 2017, 31, 503–510. [Google Scholar] [CrossRef]
- Alegría-Landa, V.; Cerroni, L.; Kutzner, H.; Requena, L. Paraprotein deposits in the skin. J. Am. Acad. Dermatol. 2017, 77, 1145–1158. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef]
- Knudsen, L.M.; Hjorth, M.; Hippe, E.; Nordic Myeloma Study Group. Renal failure in multiple myeloma: Reversibility and impact on the prognosis. Eur. J. Haematol. 2000, 65, 175–181. [Google Scholar] [CrossRef]
- Kleber, M.; Ihorst, G.; Deschler, B.; Jakob, C.; Liebisch, P.; Koch, B.; Sezer, O.; Engelhardt, M. Detection of renal impairment as one specific comorbidity factor in multiple myeloma: Multicenter study in 198 consecutive patients. Eur. J. Haematol. 2009, 83, 519–527. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Kyle, R.; Fermand, J.-P.; Rajkumar, S.V.; Miguel, J.S.; Chanan-Khan, A.; Ludwig, H.; Joshua, D.; Mehta, J.; Gertz, M.; et al. Consensus recommendations for standard investigative workup: Report of the International Myeloma Workshop Consensus. Blood 2011, 117, 4701–4705. [Google Scholar] [CrossRef]
- Haynes, R.J.; Read, S.; Collins, G.; Darby, S.C.; Winearls, C.G. Presentation and survival of patients with severe acute kidney injury and multiple myeloma: A 20-year experience from a single centre. Nephrol. Dial. Transplant. 2010, 25, 419–426. [Google Scholar] [CrossRef][Green Version]
- Dimopoulos, M.A.; Roussou, M.; Gavriatopoulou, M.; Zagouri, F.; Migkou, M.; Matsouka, C.; Barbarousi, D.; Christoulas, D.; Primenou, E.; Grapsa, I.; et al. Reversibility of renal impairment in patients with multiple myeloma treated with bortezomib-based regimens: Identification of predictive factors. Clin. Lymphoma Myeloma 2009, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A.; Cockwell, P.; Stringer, S.; Bradwell, A.; Cook, M.; Gertz, M.A.; Dispenzieri, A.; Winters, J.; Kumar, S.; Rajkumar, S.V.; et al. Early reduction of serum-free light chains associates with renal recovery in myeloma kidney. J. Am. Soc. Nephrol. 2011, 22, 1129–1136. [Google Scholar] [CrossRef]
- Herrera, G.A. The kidney in plasma cell dyscrasias: A current view and a look at the future. Contrib. Nephrol. 2007, 153, 1–4. [Google Scholar] [CrossRef]
- Hutchison, C.A.; International Kidney and Monoclonal Gammopathy Research Group; Batuman, V.; Behrens, J.; Bridoux, F.; Sirac, C.; Dispenzieri, A.; Herrera, G.A.; Lachmann, H.; Sanders, P. The pathogenesis and diagnosis of acute kidney injury in multiple myeloma. Nat. Rev. Nephrol. 2011, 8, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Gertz, M.; Zeldenrust, S.; Rajkumar, S.; Dispenzieri, A.; Fervenza, F.; Kumar, S.; Lacy, M.; Lust, J.; Greipp, P.; et al. Improvement of cast nephropathy with plasma exchange depends on the diagnosis and on reduction of serum free light chains. Kidney Int. 2008, 73, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A.; Harding, S.; Hewins, P.; Mead, G.P.; Townsend, J.; Bradwell, A.; Cockwell, P. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1684–1690. [Google Scholar] [CrossRef]
- Hutchison, C.A.; International Kidney and Monoclonal Gammopathy Research Group; Bladé, J.; Cockwell, P.; Cook, M.; Drayson, M.; Fermand, J.-P.; Kastritis, E.; Kyle, R.; Leung, N.; et al. Novel approaches for reducing free light chains in patients with myeloma kidney. Nat. Rev. Nephrol. 2012, 8, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A.; Cockwell, P.; Reid, S.; Chandler, K.; Mead, G.P.; Harrison, J.; Hattersley, J.; Evans, N.D.; Chappell, M.J.; Cook, M.; et al. Efficient removal of immunoglobulin free light chains by hemodialysis for multiple myeloma:in vitroandin vivostudies. J. Am. Soc. Nephrol. 2007, 18, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Podoll, A.S.; Finkel, K.W. Treatment of acute kidney injury with cast nephropathy. Clin. Nephrol. 2014, 82, 1–6. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Bradwell, A.R.; Cook, M.; Basnayake, K.; Basu, S.; Harding, S.; Hattersley, J.; Evans, N.D.; Chappel, M.J.; Sampson, P.; et al. Treatment of acute renal failure secondary to multiple myeloma with chemotherapy and extended high cut-off hemodialysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 745–754. [Google Scholar] [CrossRef]
- Korsak, J.; Wankowicz, Z. New options of apheresis in renal diseases: How and when? Blood Purif. 2016, 41, 1–10. [Google Scholar] [CrossRef]
- Shumak, K.H.; Rock, G.A. Therapeutic plasma exchange. N. Engl. J. Med. 1984, 310, 762–771. [Google Scholar] [CrossRef]
- Clark, W.F.; Stewart, A.K.; Rock, G.A.; Sternbach, M.; Sutton, D.M.; Barrett, B.J.; Heidenheim, A.P.; Garg, A.X.; Churchill, D.N.; Canadian Apheresis Group. Plasma exchange when myeloma presents as acute renal failure: A randomized, controlled trial. Ann. Intern. Med. 2005, 143, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Gagnon, M.; Agharazii, M.; De Serres, S.A.; Desmeules, S. Effectiveness of haemodiafiltration with heat sterilized high-flux polyphenylene HF dialyzer in reducing free light chains in patients with myeloma cast nephropathy. PLoS ONE 2015, 10, e0140463. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A. Protein-leaking membranes for hemodialysis: A new class of membranes in search of an application? J. Am. Soc. Nephrol. 2005, 16, 2421–2430. [Google Scholar] [CrossRef]
- Gondouin, B.; Hutchison, C.A. High cut-off dialysis membranes: Current uses and future potential. Adv. Chronic Kidney Dis. 2011, 18, 180–187. [Google Scholar] [CrossRef]
- Bradwell, A.R.; Carr-Smith, H.D.; Mead, G.P.; Tang, L.X.; Showell, P.J.; Drayson, M.T.; Drew, R. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clin. Chem. 2001, 47, 673–680. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Harding, S.; Mead, G.; Goehl, H.; Storr, M.; Bradwell, A.; Cockwell, P. Serum free-light chain removal by high cutoff hemodialysis: Optimizing removal and supportive care. Artif. Organs 2008, 32, 910–917. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Cockwell, P.; Moroz, V.; Bradwell, A.R.; Fifer, L.; Gillmore, J.D.; Jesky, M.D.; Storr, M.; Wessels, J.; Winearls, C.G.; et al. High cutoff versus high-flux haemodialysis for myeloma cast nephropathy in patients receiving bortezomib-based chemotherapy (EuLITE): A phase 2 randomised controlled trial. Lancet Haematol. 2019, 6, e217–e228. [Google Scholar] [CrossRef]
- Fabbrini, P.; Finkel, K.; Gallieni, M.; Capasso, G.; Cavo, M.; Santoro, A.; Pasquali, S. Light chains removal by extracorporeal techniques in acute kidney injury due to multiple myeloma: A position statement of the Onconephrology Work Group of the Italian Society of Nephrology. J. Nephrol. 2016, 29, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Wolley, M.; Jardine, M.; Hutchison, C.A. Exploring the clinical relevance of providing increased removal of large middle molecules. Clin. J. Am. Soc. Nephrol. 2018, 13, 805–814. [Google Scholar] [CrossRef]
- Krishnasamy, R.; Hawley, C.M.; Jardine, M.J.; Roberts, M.A.; Cho, Y.J.; Wong, M.G.; Heath, A.; Nelson, C.L.; Sen, S.; Mount, P.F.; et al. Design and methods of the REMOVAL-HD study: A tRial evaluating mid cut-off value membrane clearance of albumin and light chains in haemodialysis patients. BMC Nephrol. 2018, 19, 89. [Google Scholar] [CrossRef]
- Grooteman, M.P.C.; van den Dorpel, M.A.; Bots, M.L.; Penne, E.L.; van der Weerd, N.C.; Mazairac, A.H.; den Hoedt, C.H.; van der Tweel, I.; Lévesque, R.; Nubé, M.J.; et al. Contrast Investigators. Effect of online hemodiafiltra-tion on all-cause mortality and cardiovascular outcomes. J. Am. Soc. Nephrol. 2012, 23, 1087–1096. [Google Scholar] [CrossRef]
- Bolasco, P.; Ghezzi, P.M.; Serra, A.; Corazza, L.; Fundoni, G.F.; Pistis, R.; Gazzanelli, L.; Piras, A.; Accalai, G.; Calvisi, L.; et al. Effects of acetate-free haemodiafiltration (HDF) with endogenous reinfusion (HFR) on cardiac troponin levels. Nephrol. Dial. Transplant. 2011, 26, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Nubé, M.J.; Grooteman, M.P.C.; Blankestijn, P. Hemodiafiltration Theory, Technology and Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2016; pp. 87–94. [Google Scholar]
- Vallée, A.G.; Chenine, L.; Leray-Moragues, H.; Patrier, L.; Cognot, C.; Cartron, G.; Cristol, J.-P.; Canaud, B. Online high-efficiency haemodiafiltration achieves higher serum free light chain removal than high-flux haemodialysis in multiple myeloma patients: Preliminary quantitative study. Nephrol. Dial. Transplant. 2011, 26, 3627–3633. [Google Scholar] [CrossRef]
- Bourguignon, C.; Chenine, L.; Bargnoux, A.S.; Leray-Moragues, H.; Canaud, B.; Cristol, J.-P.; Morena, M. Hemodiafiltration improves free light chain removal and normalizes κ/λ ratio in hemodialysis patients. J. Nephrol. 2016, 29, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Wratten, M.L.; Ghezzi, P.M. Hemodiafiltration with endogenous reinfusion. Contrib. Nephrol. 2007, 158, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, S.; Iannuzzella, F.; Corradini, M.; Mattei, S.; Bovino, A.; Stefani, A.; Palladino, G.; Caiazzo, M. A novel option for reducing free light chains in myeloma kidney: Supra-hemodiafiltration with endogenous reinfusion (HFR). J. Nephrol. 2015, 28, 251–254. [Google Scholar] [CrossRef]
- Menè, P.; Giammarioli, E.; Fofi, C.; Antolino, G.; La Verde, G.; Tafuri, A.; Punzo, G.; Festuccia, F. Serum free light chains removal by HFR hemodiafiltration in patients with multiple myeloma and acute kidney injury: A case series. Kidney Blood Press. Res. 2018, 43, 1263–1272. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Patient | Age | M/F | eGFR, mL/min | uProt, g/day | Diagnosis |
|---|---|---|---|---|---|
| M.M.B. | 72 | F | 10.6 (pre-HD) | 5.0 | AKI, AL Amyloidosis, IgG λ |
| R.D.C. | 72 | F | 9.5 (pre-HD) | 7.5 | AKI, AL Amyloidosis, IgG λ |
| B.M. | 73 | F | 11.9 (pre-HD) | 5.0 | AKI, Cast nephropathy, IgG k |
| A.P. | 76 | M | 12.4 (pre-HD) | 10.0 | AKI, Cast nephr., AL Amyl., IgG λ |
| 73.2 ± 1.9 | 11.1 ± 1.3 | 6.9 ± 2.4 |
| Patient | Age | M/F | eGFR, mL/min | uProt, g/day | Diagnosis |
|---|---|---|---|---|---|
| G.C. | 65 | M | 33.1 | 5.2 | LCDD, IgM λ |
| E.D.B. | 72 | M | 30.4 | 1.1 | LCDD, IgM λ |
| L.F. | 66 | F | 25.1 | 8.8 | LCDD, IgG k |
| I.D.T. | 71 | F | 50.7 | 13.2 | LCDD, IgM λ |
| G.G. | 60 | F | 32.3 | 4.1 | LCDD, IgG k |
| M.L. | 45 | M | 96.7 | 8.8 | LCDD, IgG k |
| G.M.M. | 42 | F | 107.1 | 2.2 | LCDD, IgG k |
| M.C.S. | 52 | F | 65.3 | 5.8 | LCDD, IgG k MM |
| D.S. | 64 | M | 45.8 | 8.5 | LCDD, IgG k MM |
| L.B.N. | 51 | F | 36.6 | 1.9 | AL Amyloidosis, IgG λ mMM |
| V.T. | 62 | M | 26.3 | 1.2 | AL Amyloidosis, IgA λ |
| T.A. | 64 | M | 52.2 | 11 | AL Amyloidosis, IgA λ mMM |
| G.C. | 54 | M | 108.5 | 5 | AL Amyloidosis + FibGNF, IgM l |
| R.C. | 68 | M | 100 | 4.8 | AL Amyloidosis, IgA λ |
| R.G. | 54 | F | 160.2 | 3.1 | AL Amyloidosis, IgM λ MM |
| M.L. | 59 | M | 94 | 5.3 | AL Amyloidosis, IgM λ |
| E.L.C. | 70 | M | 84.3 | 4.6 | AL Amyl., B-cell lymphoma, IgG k |
| P.L. | 70 | F | 90.1 | 5.2 | AL Amyloidosis, IgM λ |
| E.P. | 54 | F | 120.5 | 4.4 | AL Amyloidosis, IgM λ |
| F.P. | 66 | M | 39.4 | 7.2 | AL Amyloidosis, IgM λ MM |
| 60.45 ± 8.76 | 69.93 ± 38.36 | 5.57 ± 3.20 |
| Major Regimens/Associations | Renal Toxicities |
|---|---|
| Alkylating agents | |
| Melphalan | AKI |
| Cisplatin | ATN > Fanconi syndrome > TMA |
| Cyclophosphamide | Haemorrhagic cystitis, SIAD |
| Anthracyclines | Glomerulonephritis, proteinuria |
| Proteasome inhibitors | |
| Bortezomib | TMA > interstitial nephritis |
| Carfilzomib | TMA > hypertension (vasoconstrictor effects) |
| Immunomodulators | |
| Thalidomide | ↑ serum creatinine |
| Lenalidomide | variable, ↑ serum creatinine, hypokalemia |
| Pomalidomide | AKI, crystal nephropathy |
| BRAF inhibitors | |
| Vemurafenib | AKI > interstitial nephritis |
| Dabrafenib | AKI > interstitial nephritis |
| SLAMF7 antagonists | |
| Elotuzumab | AKI |
| AKT/MTOR inhibitors | |
| Perifosine | Hypophosphatemia |
| Rapamycine | Proteinuria, rare AKI |
| Everolimus | Proteinuria, rare AKI |
| Anti-IL6 MAb | |
| Siltuximab | Hyperuricemia, hyperkalemia |
| Anti PD-1 immune checkpoint inhibitors | |
| Nivolumab | AKI > interstitial nephritis |
| Pembrolizumab | AKI > Interstitial nephritis |
| Anti-KIR agents | |
| Lirilumab | AKI, hyperuricemia |
| Kappa FLC (mg/dL) | Lambda FLC (mg/dL) | |
|---|---|---|
| Baseline | 5599.6 ± 672.0 | 4176.8 ± 279.0 |
| After HFR | 2987.1 ± 515.1 | 1603.2 ± 151.8 |
| n of patients | 3 | 2 |
| % removal | 46.5 ± 4.5 | 60.2 ± 19.0 |
| p | <0.002 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menè, P.; Moioli, A.; Stoppacciaro, A.; Lai, S.; Festuccia, F. Acute Kidney Injury in Monoclonal Gammopathies. J. Clin. Med. 2021, 10, 3871. https://doi.org/10.3390/jcm10173871
Menè P, Moioli A, Stoppacciaro A, Lai S, Festuccia F. Acute Kidney Injury in Monoclonal Gammopathies. Journal of Clinical Medicine. 2021; 10(17):3871. https://doi.org/10.3390/jcm10173871
Chicago/Turabian StyleMenè, Paolo, Alessandra Moioli, Antonella Stoppacciaro, Silvia Lai, and Francescaromana Festuccia. 2021. "Acute Kidney Injury in Monoclonal Gammopathies" Journal of Clinical Medicine 10, no. 17: 3871. https://doi.org/10.3390/jcm10173871
APA StyleMenè, P., Moioli, A., Stoppacciaro, A., Lai, S., & Festuccia, F. (2021). Acute Kidney Injury in Monoclonal Gammopathies. Journal of Clinical Medicine, 10(17), 3871. https://doi.org/10.3390/jcm10173871