
Role of Advanced Glycation End-Products and Other Ligands for AGE Receptors in Thyroid Cancer Progression

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
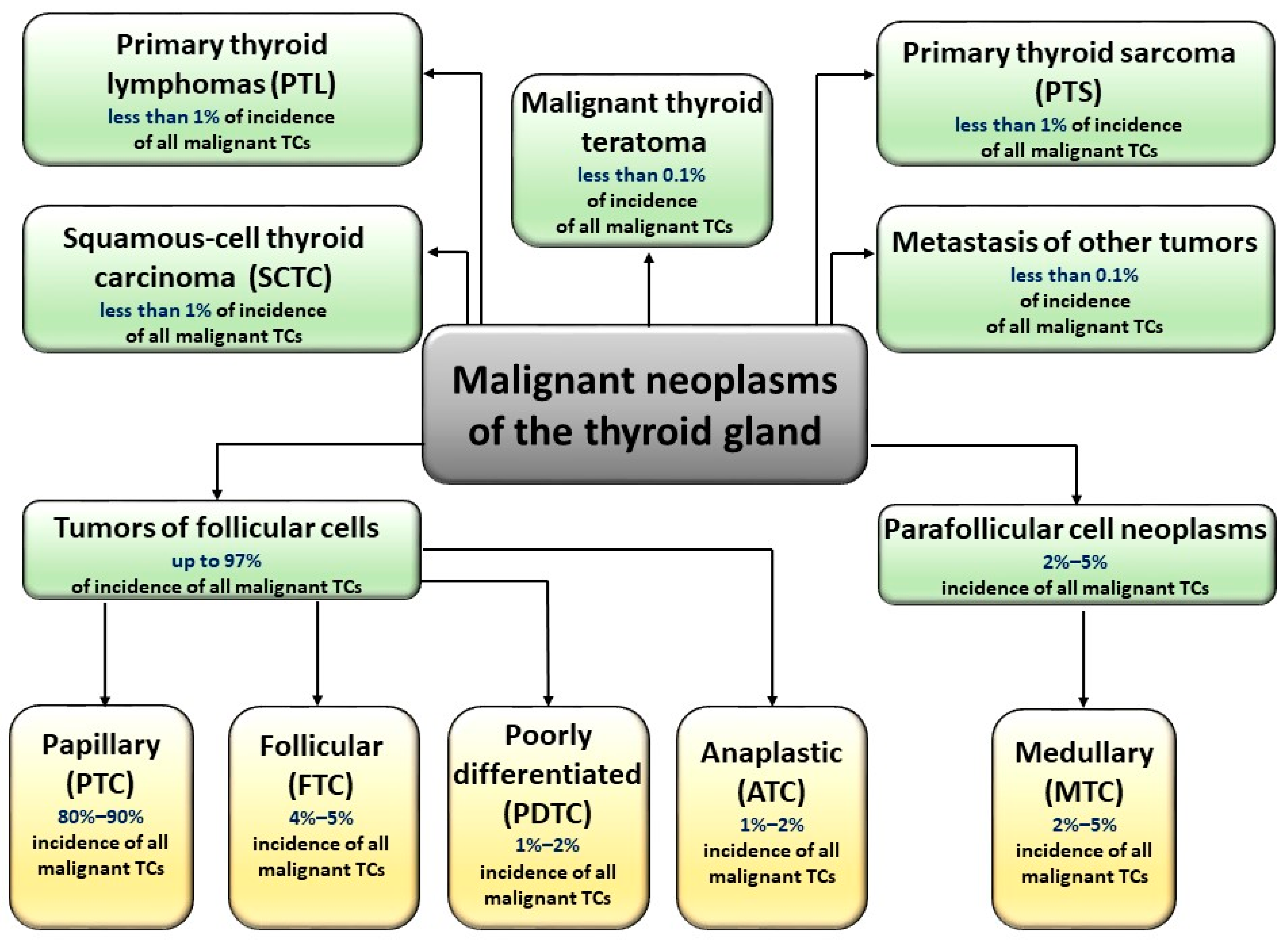
2. Advanced Glycation End-Products and Their Possible Linkage with Thyroid Cancers
2.1. The Glycation Process, Advanced Glycation End-Products, and Their Receptors
2.2. AGEs, RAGE, and Possible Therapeutic Application of RAGE Antagonists in the State of Carcinogenesis
3. AGEs as a Possible Link between Oxidative Stress and the Incidence of TCs
3.1. Insights on Carcinogenesis in Context of Glycation, Inflammation, Oxidative/Carbonyl/Nitrosative Stress, and Their Impact on Cell Integrity and Function
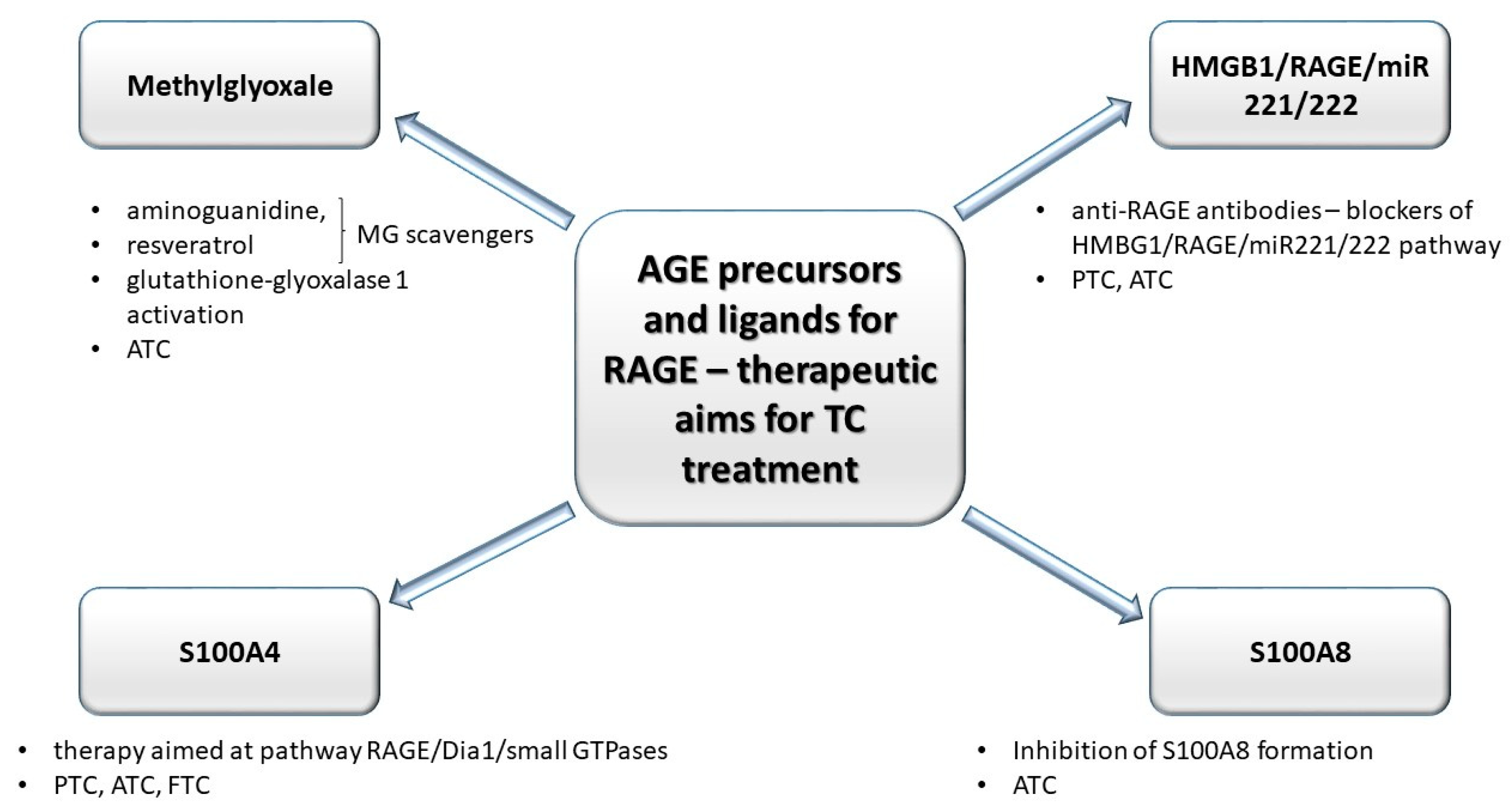
3.2. Selected Advanced Glycation End-Product Precursors and RAGE Ligands as Factors of Thyroid Tumor Progression. Anti-Glycative Treatment as a Potentially-Promising Tool in Future Thyroid Cancer Therapy
3.2.1. Methylglyoxal
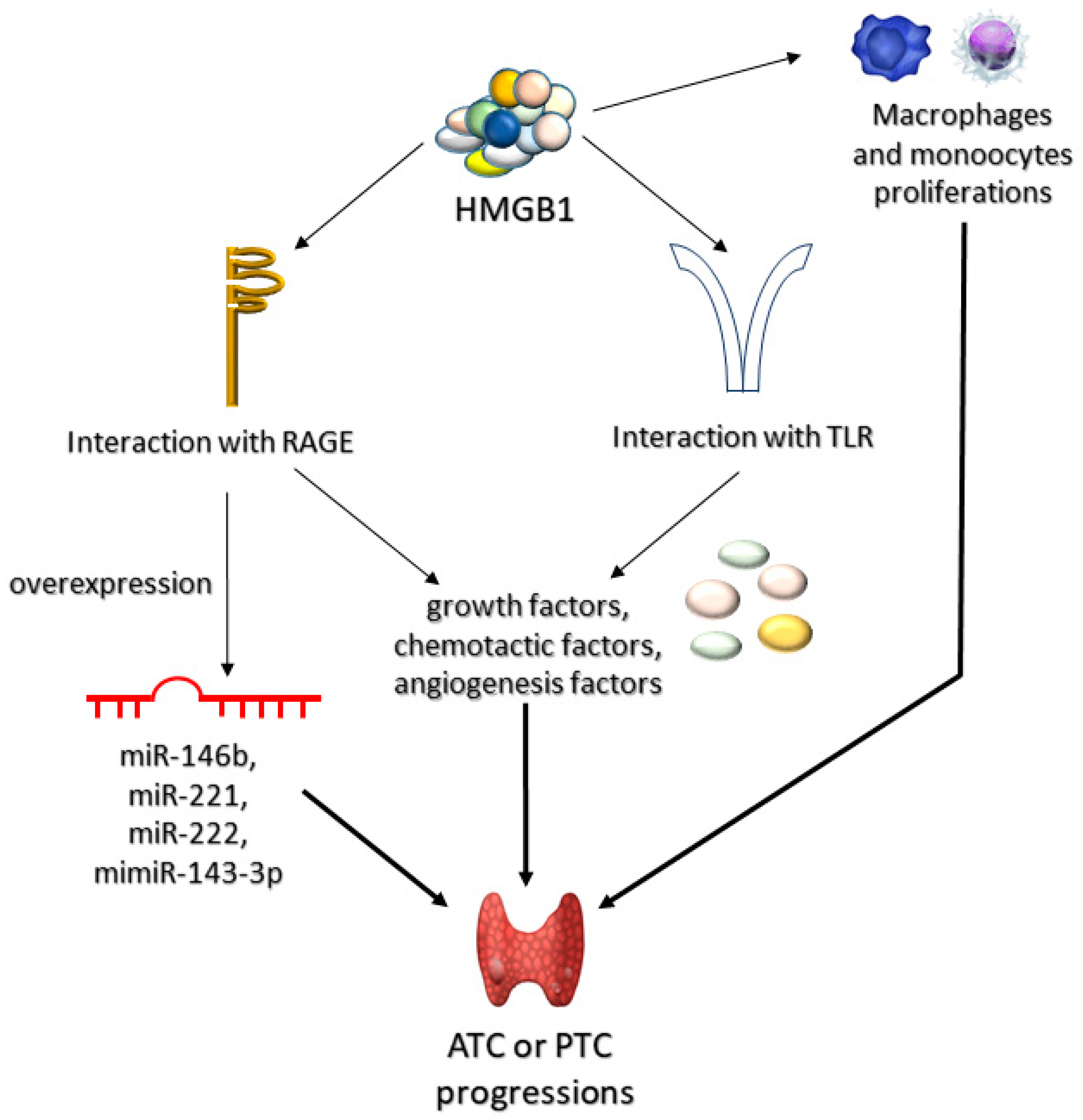
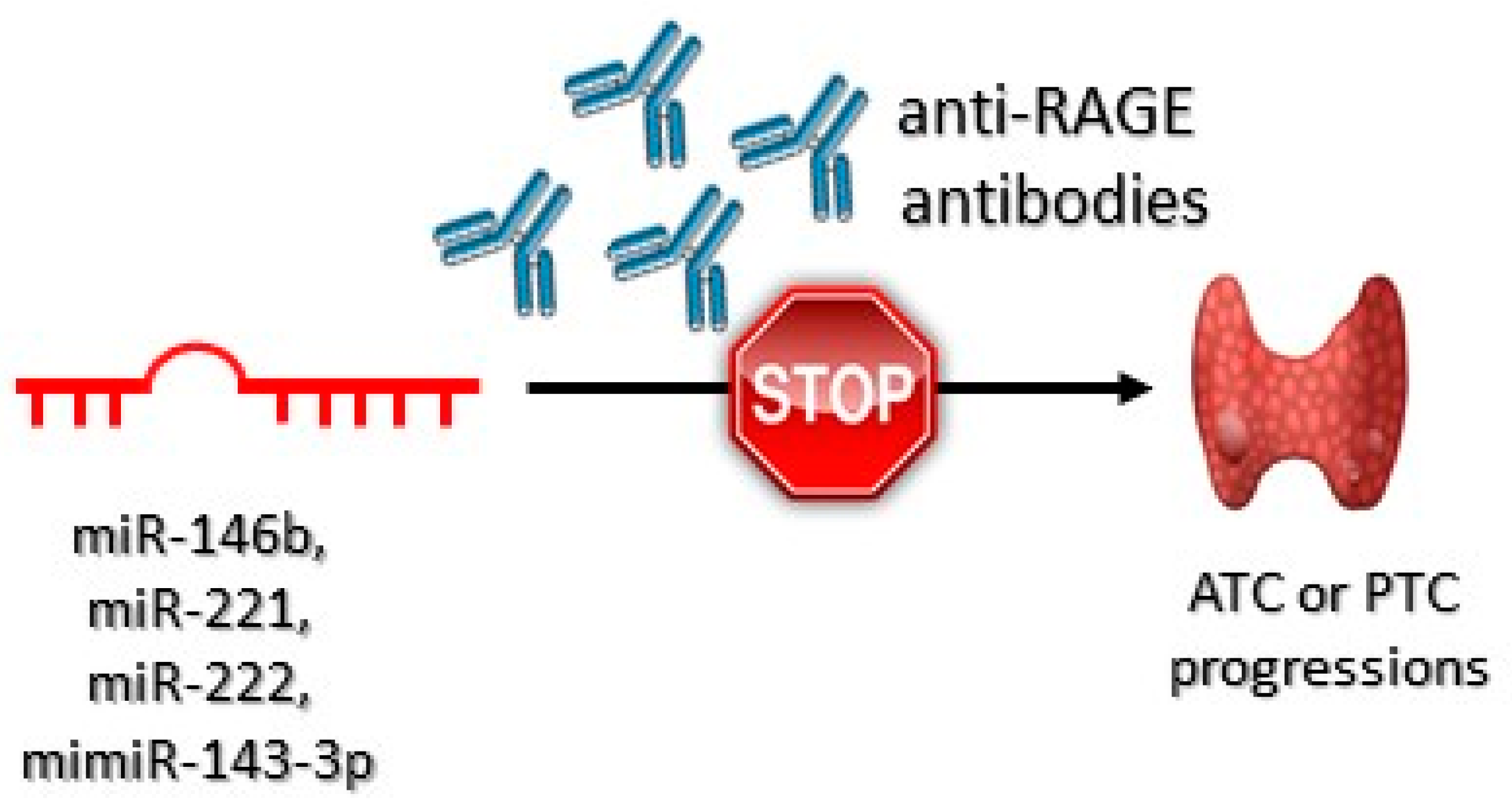
3.2.2. High-Mobility Group Box 1 Protein (HMGB1)
3.2.3. S100 Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chmielik, E.; Rusinek, D.; Oczko-Wojciechowska, M.; Jarzab, M.; Krajewska, J.; Czarniecka, A.; Jarzab, B. Heterogeneity of thyroid cancer. Pathobiology 2018, 85, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Gundara, J.S.; Glover, A.; Serpell, J.; Sidhu, S.B. MicroRNA Expression Profiles in the management of papillary thyroid cancer. Oncologist 2014, 19, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Kakudo, K.; Bychkov, A.; Bai, Y.; Li, Y.; Liu, Z.; Jung, C.K. The new 4th edition World Health Organization classification for thyroid tumors, Asian perspectives. Pathol. Int. 2018, 68, 641–664. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.H.; Cao, J.; Wang, J.Y.; Huang, Y.Q.; Lan, X.B.; Yu, B.; Wen, Q.L.; Cai, X.J. Nomograms predicting disease-specific regional recurrence and distant recurrence of papillary thyroid carcinoma following partial or total thyroidectomy. Medicine 2017, 96, e7575. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, M. Screening for high blood pressure in children and adolescents: U.S. preventive services task force recommendation statement. Am. Fam. Physician 2021, 103, 239A–239D, Online. [Google Scholar]
- Clarke, C.A.; Reynolds, P.; Oakley-Girvan, I.; Lee, E.; Lu, Y.; Yang, J.; Moy, L.M.; Bernstein, L.; Horn-Ross, P.L. Indicators of microbial-rich environments and the development of papillary thyroid cancer in the California Teachers Study. Cancer Epidemiol. 2015, 39, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 Pt 24, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Azouzi, N.; Cailloux, J.; Cazarin, J.M.; Knauf, J.A.; Cracchiolo, J.; Ghuzian, A.A.; Hartl, D.; Polak, M.; Carre, A.; Mzibir, M.E.; et al. NADPH Oxidase NOX4 is a critical mediator of BRAFV600E-induced downregulation of the sodium/iodide symporter in papillary thyroid carcinomas. Antioxid. Redox Signal. 2017, 26, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Marotta, V.; Sciammarella, C.; Colao, A.; Faggiano, A. Application of molecular biology of differentiated thyroid cancer for clinical prognostication. Endocr. Relat. Cancer 2016, 23, R499–R515. [Google Scholar] [CrossRef] [Green Version]
- Marotta, V.; Malandrino, P.; Russo, M.; Panariello, I.; Ionna, F.; Chiofalo, M.G.; Pezzullo, L. Fathoming the link between anthropogenic chemical contamination and thyroid cancer. Crit. Rev. Oncol. Hematol. 2020, 150, 102950. [Google Scholar] [CrossRef]
- Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Pacini, F.; Schlumberger, M.; et al. Revised American thyroid association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.G.; Carty, S.E.; McCoy, K.L.; Ohori, N.P.; LeBeau, S.O.; Seethala, R.R.; Nikiforova, M.N.; Nikiforov, Y.E.; Yip, L. Preoperative detection of RAS mutation may guide extent of thyroidectomy. Surgery 2017, 161, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, M.R.; Gharib, H. Continuing controversies in the management of thyroid nodules. Ann. Intern. Med. 2005, 142, 926–931. [Google Scholar] [CrossRef]
- Guerra, A.; Di Stasi, V.; Zeppa, P.; Faggiano, A.; Marotta, V.; Vitale, M. BRAF(V600E) assessment by pyrosequencing in fine needle aspirates of thyroid nodules with concurrent Hashimoto’s thyroiditis is a reliable assay. Endocrine 2014, 45, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Marotta, V.; Bifulco, M.; Vitale, M. Significance of RAS mutations in thyroid benign nodules and non-medullary thyroid cancer. Cancers 2021, 13, 3785. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Chia, S.Y. Circulating thyroid cancer markers. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 383–388. [Google Scholar] [CrossRef]
- Tallini, G.; Ghossein, R.A.; Emanuel, J.; Gill, J.; Kinder, B.; Dimich, A.B.; Costa, J.; Robbins, R.; Burrow, G.N.; Rosai, J. Detection of thyroglobulin, thyroid peroxidase, and RET/PTC1 mRNA transcripts in the peripheral blood of patients with thyroid disease. J. Clin. Oncol. 1998, 16, 1158–1166. [Google Scholar] [CrossRef]
- Ditkoff, B.A.; Marvin, M.R.; Yemul, S.; Shi, Y.J.; Chabot, J.; Feind, C.; Lo Gerfo, P.; Clark, O.H.; McHenry, C.R. Detection of circulating thyroid cells in peripheral blood. Surgery 1996, 120, 959–965. [Google Scholar] [CrossRef]
- Span, P.N.; Sleegers, M.J.M.; Van den Broek, W.J.; Ross, H.A.; Nieuwlaat, W.A.; Hermus, A.R.M.M.; Sweep, C.G.J. Quantitative detection of peripheral thyroglobulin mRNA has limited clinical value in the follow-up of thyroid cancer patients. Ann. Clin. Biochem. 2003, 40, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Bugalho, M.J.; Domingues, R.S.; Pinto, A.C.; Garrão, A.; Catarino, A.L.; Ferreira, T.; Limbert, E.; Sobrinho, L. Detection of thyroglobulin mRNA transcripts in peripheral blood of individuals with and without thyroid glands: Evidence for thyroglobulin expression by blood cells. Eur. J. Endocrinol. 2001, 145, 409–413. [Google Scholar] [CrossRef]
- Bojunga, J.; Röddiger, S.; Stanisch, M.; Kusterer, K.; Kurek, R.; Renneberg, H.; Adams, S.; Lindhorst, E.; Usadel, K.H.; Schumm-Draeger, P.M. Molecular detection of thyroglobulin mRNA transcripts in peripheral blood of patients with thyroid disease by RT-PCR. Br. J. Cancer 2000, 82, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugazzola, L.; Mihalich, A.; Persani, L.; Cerutti, N.; Reina, M.; Bonomi, M.; Ponti, E.; Mannavola, D.; Giammona, E.; Vannucchi, G.; et al. Highly sensitive serum thyroglobulin and circulating thyroglobulin mRNA evaluations in the management of patients with differentiated thyroid cancer in apparent remission. J. Clin. Endocrinol. Metab. 2002, 87, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Grammatopoulos, D.; Elliott, Y.; Smith, S.C.; Brown, I.; Grieve, R.J.; Hillhouse, E.W.; Levine, M.A.; Ringel, M.D. Measurement of thyroglobulin mRNA in peripheral blood as an adjunctive test for monitoring thyroid cancer. J. Clin. Pathol. Mol. Pathol. 2003, 56, 162–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.; Arciaga, R.; Siperstein, A.; Milas, M.; Warshawsky, I.; Reddy, S.S.K.; Gupta, M.K. Rapid communication: Thyrotropin receptor/thyroglobulin messenger ribonucleic acid in peripheral blood and fine-needle aspiration cytology: Diagnostic synergy for detecting thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 1921–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milas, M.; Mazzaglia, P.; Chia, S.Y.; Skugor, M.; Berber, E.; Reddy, S.; Gupta, M.; Siperstein, A. The utility of peripheral thyrotropin mRNA in the diagnosis of follicular neoplasms and surveillance of thyroid cancers. Surgery 2007, 141, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Boscaro, M.; Pacenti, M.; Taccaliti, A.; Palù, G. Evaluation of circulating thyroid-specific transcripts as markers of thyroid cancer relapse. Int. J. Cancer 2004, 110, 914–920. [Google Scholar] [CrossRef]
- Roddiger, S.J.; Bojunga, J.; Klee, V.; Stanisch, M.; Renneberg, H.; Lindhorst, E.; Usadel, K.H.; Kusterer, K.; Schumm-Draeger, P.M.; Kurek, R. Detection of thyroid peroxidase mRNA in peripheral blood of patients with malignant and benign thyroid diseases. J. Mol. Endocrinol. 2002, 29, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Endo, T.; Kawaguchi, A.; Ikeda, M.; Katoh, R.; Kawaoi, A.; Muramatsu, A.; Onaya, T. Increased expression of the sodium/iodide symporter in papillary thyroid carcinomas. J. Clin. Investig. 1998, 101, 1296–1300. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.F.; Giuffrida, D.; Schlumberger, M.; Filetti, S. Sodium-iodide symporter (NIS) gene expression in lymph-node metastases of papillary thyroid carcinomas. Eur. J. Endocrinol. 2000, 143, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Beer, J.C.; Liebenberg, L. Does cancer risk increase with HbA1c, independent of diabetes? Br. J. Cancer 2014, 110, 2361–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Choudhuri, S.; Dutta, D.; Chowdhury, I.H.; Mitra, B.; Sen, A.; Mandal, L.K.; Mukhopadhyay, S.; Bhattacharya, B. Association of hyperglycemia mediated increased advanced glycation and erythrocyte antioxidant enzyme activity in different stages of diabetic retinopathy. Diabetes Res. Clin. Pract. 2013, 100, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, G.; Lakshmanan, D.K.; Raju, K.; Elangovan, A.; Nambirajan, G.; Devanesan, A.A.; Thilagar, S. Food advanced glycation end products as potential endocrine disruptors: An emerging threat to contemporary and future generation. Environ. Int. 2019, 123, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Kamil, M.; Hasan, A.; Irfan, S.; Sheikh, S.; Khatoon, A.; Nazir, A.; Mir, S.S. Advanced glycation end products (AGEs), protein aggregation and their cross talk: New insight in tumorigenesis. Glycobiology 2019, 30, 49–57. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [Green Version]
- McCreery, M.Q.; Balmain, A. Chemical carcinogenesis models of cancer: Back to the future. Annu. Rev. Cancer Biol. 2017, 1, 295–312. [Google Scholar] [CrossRef]
- Xing, M. BRAF mutation in thyroid cancer. Endocr.-Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Santisteban, P. New insights in thyroid follicular cell biology and its impact in thyroid cancer therapy. Endocr.-Relat. Cancer 2007, 14, 957–977. [Google Scholar] [CrossRef]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucera, C.; Lawler, J.; Hodin, R.; Parangi, S. The BRAFV600E mutation: What is it really orchestrating in thyroid cancer? Oncotarget 2010, 1, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, Y.; Tsuij, E.; Yagi, K.; Matsusaka, K.; Tsuji, S.; Kurebayashi, J.; Ogawa, T.; Aburatani, H.; Kaneda, A. Aberrantly methylated genes in human papillary thyroid cancer and their association with BRAF/RAS mutation. Front. Gen. 2013, 4, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Boelaert, K. Thyroid gland: Revised guidelines for the management of thyroid cancer. Nat. Rev. Endocrinol. 2010, 6, 185–186. [Google Scholar] [CrossRef]
- Morgan, P.E.; Dean, R.T.; Davies, M.J. Inactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation products. Arch. Biochem. Biophys. 2002, 403, 259–269. [Google Scholar] [CrossRef]
- Kandaraki, E.A.; Chatzigeorgiou, A.; Papageorgiou, E.; Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G.; Koutsilieris, M.; Diamanti-Kandarakis, E. Advanced glycation end products interfere in luteinizing hormone and follicle stimulating hormone signaling in human granulosa KGN cells. Exp. Biol. Med. 2018, 243, 29–33. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Thorpe, S.R.; Fu, M.X.; Harper, C.M.; Yoo, J.; Kim, S.M.; Wong, H.; Peters, A.L. Glycation impairs high-density lipoprotein function. Diabetologia 2000, 43, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef]
- Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Glycation & the RAGE axis: Targeting signal transduction through DIAPH1. Expert Rev. Proteom. 2017, 14, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Berning, K.; Quan, C.; Zhang, Y.T. Glycation of antibodies: Modification, methods and potential effects on biological functions. MAbs 2017, 9, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gstöttner, C.; Reusch, D.; Haberger, M.; Dragan, I.; Van Veelen, P.; Kilgour, D.P.A.; Tsybin, Y.O.; van der Burgt, Y.E.M.; Wuhrer, M.; Nicolardi, S. Monitoring glycation levels of a bispecific monoclonal antibody at subunit level by ultrahigh-resolution MALDI FT-ICR mass spectrometry. MAbs 2020, 12, 1682403. [Google Scholar] [CrossRef] [Green Version]
- Murata, M. Browning and pigmentation in food through the Maillard reaction. Glycoconj. J. 2020, 38, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, M.; Bronowicka-Szydełko, A.; Gostomska-Pampuch, K.; Szkudlarek, J.; Bartyś, A.; Bieg, T.; Gamian, E.; Kochman, A.; Picur, B.; Pietkiewicz, J.; et al. The melibiose-derived glycation product mimics a unique epitope present in human and animal tissues. Sci. Rep. 2021, 11, 2940. [Google Scholar] [CrossRef]
- Baynes, J.W. The role of AGEs in aging: Causation or correlation. Exp. Gerontol. 2001, 36, 1527–1537. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Salvayre, R.; Augé, N.; Pamplona, R.; Portero-Otín, M. Hyperglycemia and glycation in diabetic complications. Antioxid. Redox Signal. 2009, 11, 3071–3109. [Google Scholar] [CrossRef] [PubMed]
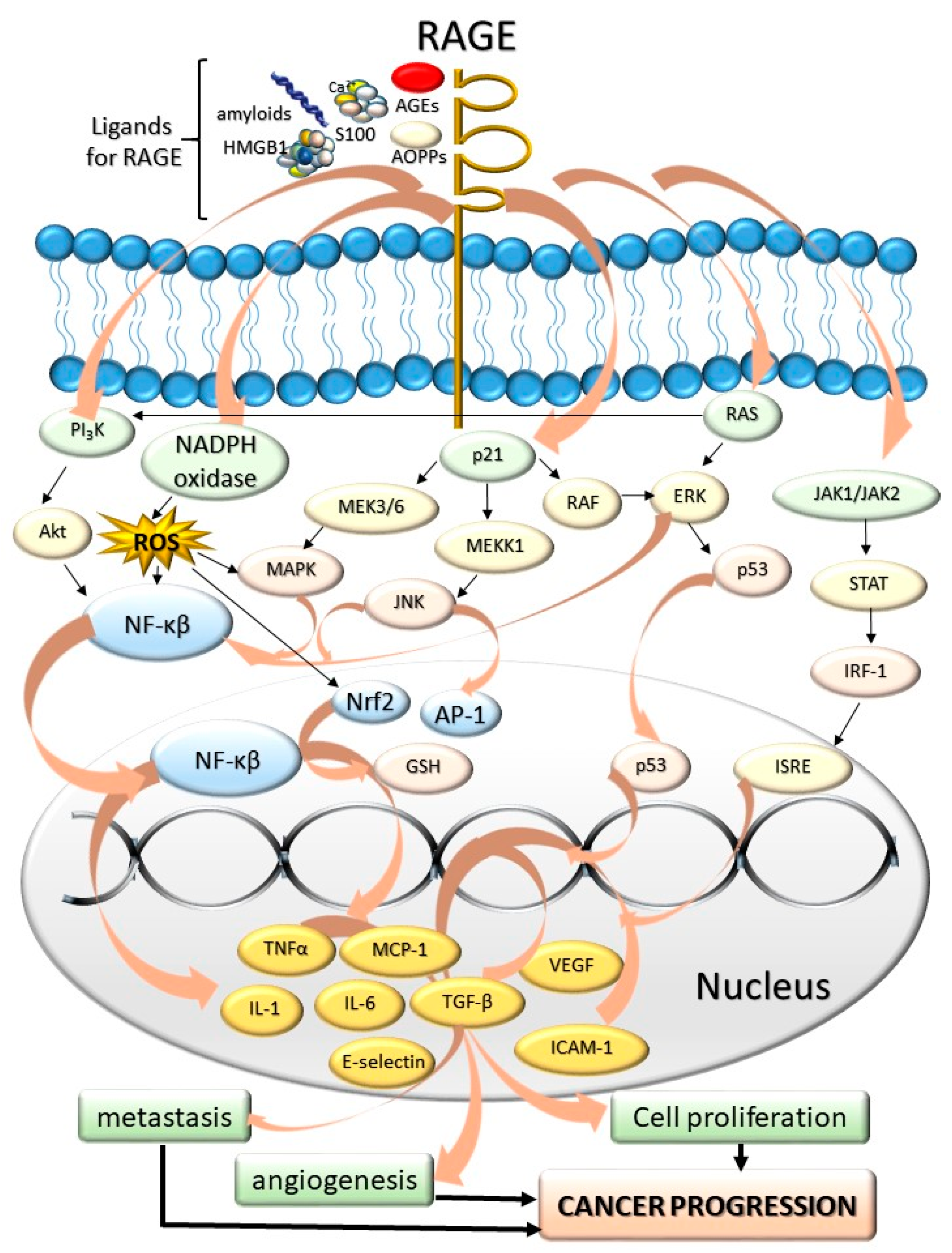
- Palanissami, G.; Paul, S.F.D. RAGE and its ligands: Molecular interplay between glycation, inflammation, and hallmarks of cancer—A review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Zahng, W.; Randell, E.W.; Sun, G.; Likhodii, S.; Liu, M.; Furey, A.; Zhai, G. Hyperglycemia-related advanced glycation end-products is associated with the altered phosphatidylcholine metabolism in osteoarthritis patients with diabetes. PLoS ONE 2017, 12, e0184105. [Google Scholar] [CrossRef] [Green Version]
- Saheem, A.; Mohd, Y.K.; Zeeshan, R.; Hamda, K.; Zeba, S.; Shahnawaz, R.; Uzma, S.; Mohd, S.K.; Saeed, M.; Sultan, A.; et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Ionta, V.; Blasetti Fantauzzi, C.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. The advanced glycation end-product Nϵ-carboxymethyllysine promotes progression of pancreatic cancer: Implications for diabetes-associated risk and its prevention. J. Pathol. 2018, 245, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Konopka, C.J.; Woźniak, M.; Hedhli, J.; Siekierzycka, A.; Skokowski, J.; Pęksa, R.; Matuszewski, M.; Munirathinam, G.; Kajdacsy-Balla, A.; Dobrucki, I.T.; et al. Quantitative imaging of the receptor for advanced glycation end-products in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2562–2576. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sharma, V.R.; Gupta, G.K.; Ashraf, G.M.; Kamal, M.A. Advanced Glycation End Products (AGEs), Glutathione and Breast Cancer: Factors, Mechanism and Therapeutic Interventions. Curr. Drug Metab. 2019, 20, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Derk, J.; MacLean, M.; Juranek, J.; Schmidt, A.M. The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration. J. Alzheimer’s Dis. Park. 2018, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Melnikova, A.K.; Seferbekova, Z.N.; Barinova, K.V.; Schmalhausen, E.V. Glycation, glycolysis, and neurodegenerative diseases: Is there any connection? Biochemistry 2017, 82, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Kitzmiller, J.L.; Ferrara, A.; Peng, T.; Cissell, M.A.; Kim, C.; Cowie, C.C.; Casagrande, S.S.; Menke, A.; Cissell, M.A.; Eberhardt, M.S.; et al. Association of RAGE with acute ischemic stroke prognosis in type 2 diabetes. Ir. J. Med. Sci. 2021, 190, 625–630. [Google Scholar] [CrossRef]
- Nakhjavani, M.R.J.; Jafarpour, M.; Ghorbanihaghjo, A.; Azar, S.A.; Mahdavi, A.M. Relationship between serum-soluble receptor for advanced glycation end products (sRAGE) and disease activity in rheumatoid arthritis patients. Mod. Rheumatol. 2019, 29, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Khan, M.S.; Akhter, F.; Husain, F.M.; Ahmad, S.; Chen, L. The non-enzymatic glycation of LDL proteins results in biochemical alterations—A correlation study of Apo B 100-AGE with obesity and rheumatoid arthritis. Int. J. Biol. Macromol. 2019, 122, 195–200. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for advanced glycation end products (RAGE) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: Insights from human subjects and animal models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced glycation end products: Potential mechanism and therapeutic target in cardiovascular complications under diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation reduces the stability of apoai and increases HDL dysfunction in diet-controlled type 2 diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Nedić, O.; Rattan, S.I.S.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47, 28–38. [Google Scholar] [CrossRef]
- Lan, K.C.; Chiu, C.Y.; Kao, C.W.; Huang, K.H.; Wang, C.C.; Huang, K.T.; Tsai, K.S.; Sheu, M.L.; Liu, S.H. Advanced glycation end-products induce apoptosis in pancreatic islet endothelial cells via NF-κB-activated cyclooxygenase-2/prostaglandin e2 up-regulation. PLoS ONE 2015, 10, e0124418. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, S.; Mao, X.; Wang, W.; Tai, H. Inhibition effect of curcumin on TNF-α and MMP-13 expression induced by advanced glycation end products in chondrocytes. Pharmacology 2013, 91, 77–85. [Google Scholar] [CrossRef]
- Polykretis, P.; Luchinat, E.; Boscaro, F.; Banci, L. Methylglyoxal interaction with superoxide dismutase 1. Redox Biol. 2020, 30, 01421. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Zhang, M.; Huang, K.; Yao, Z.; Rao, P.; Cai, X.; Xiao, J. Advanced glycation end products inhibit the osteogenic differentiation potential of adipose-derived stem cells by modulating Wnt/β-catenin signalling pathway via DNA methylation. Cell Prolif. 2020, 53, e12834. [Google Scholar] [CrossRef] [PubMed]
- Sell, D.R.; Monnier, V.M. Molecular basis of arterial stiffening: Role of glycation-a mini-review. Gerontology 2012, 58, 227–237. [Google Scholar] [CrossRef]
- Dhama, K.; Latheef, S.K.; Dadar, M.; Samad, H.A.; Munjal, A.; Khandia, R.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Bhatt, P.; et al. Biomarkers in stress related diseases/disorders: Diagnostic, prognostic, and therapeutic values. Front. Mol. Biosci. 2019, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- McRobert, A.E.; Gallicchio, M.; Jerums, G.; Cooper, M.E.; Bach, L.A. The amino-terminal domains of the ezrin, radixin, and moesin (ERM) proteins bind advanced glycation end products, an interaction that may play a role in the development of diabetic complications. J. Biol. Chem. 2003, 278, 25783–25789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Fournet, M.; Bonté, F.; Desmoulière, A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis. 2018, 9, 880–900. [Google Scholar] [CrossRef] [Green Version]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Teodorowicz, M.; Hendriks, W.H.; Wichers, H.J.; Savelkoul, H.F.J. Immunomodulation by processed animal feed: The role of maillard reaction products and advanced glycation end-products (AGEs). Front. Immunol. 2018, 9, 2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, K. Is there any evidence that AGE/sRAGE is a universal biomarker/risk marker for diseases? Mol. Cell. Biochem. 2019, 451, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Stratmann, B.; Ruetter, R.; Buenting, C.E.; Menart, B.; Weiss, J.; Vlassara, H.; Koschinsky, T.; Tschoepe, D. Advanced glycation end products strongly activate platelets. Eur. J. Nutr. 2009, 48, 475–481. [Google Scholar] [CrossRef] [PubMed]
- de Laat, M.A.; Kyaw-Tanner, M.T.; Sillence, M.N.; McGowan, C.M.; Pollitt, C.C. Advanced glycation end-products in horses with insulin-induced laminitis. Vet. Immunol. Immunopathol. 2012, 145, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sick, E.; Brehin, S.; André, P.; Coupin, G.; Landry, Y.; Takeda, K.; Gies, J.P. Advanced glycation end products (AGEs) activate mast cells. Br. J. Pharmacol. 2010, 161, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Heidari, F.; Rabizadeh, S.; Ali Mansournia, M.; Mirmiranpoor, H.; Salehi, S.S.; Akhavan, S.; Esteghamati, A.; Nakhjavani, M. Inflammatory, oxidative stress and anti-oxidative markers in patients with endometrial carcinoma and diabetes. Cytokine 2019, 120, 186–190. [Google Scholar] [CrossRef]
- Mollace, M.; Coluccio, M.L.; Donato, G.; Mollace, V.; Malara, N. Cross-talks in colon cancer between RAGE/AGEs axis and inflammation/immunotherapy. Oncotarget 2021, 12, 1281–1295. [Google Scholar] [CrossRef]
- Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [CrossRef] [Green Version]
- Ruggeri, R.M.; Barbalace, M.C.; Cristani, M.T.; Alibrandi, A.; Giovinazzo, S.; Giuffrida, G.; Trimarchi, F.; Cannavò, S.; Campennì, A. Serum levels of advanced glycation end products (AGEs) are increased and their soluble receptor (sRAGE) reduced in Hashimoto’s thyroiditis. J. Endocrinol. Investig. 2020, 43, 1337–1342. [Google Scholar] [CrossRef]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Origlia, N.; Righi, M.; Capsoni, S.; Cattaneo, A.; Fang, F.; Stern, D.M.; Chen, J.C.; Schmidt, A.M.; Arancio, O.; Yan, S.D.; et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J. Neurosci. 2008, 28, 3521–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, A.; Delgado-López, F.; González, I.; Pérez-Castro, R.; Romero, J.; Rojas, I. The receptor for advanced glycation end-products: A complex signaling scenario for a promiscuous receptor. Cell. Signal. 2013, 25, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Guh, J.Y.; Hung, W.C.; Yang, M.L.; Lai, Y.H.; Chen, H.C.; Chuang, L.Y. Role of the Janus kinase (JAK)/signal transducters and activators of transcription (STAT) cascade in advanced glycation end-product-induced cellular mitogenesis in NRK-49F cells. Biochem. J. 1999, 342, 231–238. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [Green Version]
- Qian, Q.Q.; Zhang, X.; Wang, Y.W.; Xu, J.W.; Dong, H.Q.; Li, N.N.; Qian, Y.N.; Gui, B. Pro-inflammatory role of high-mobility group box-1 on brain mast cells via the RAGE/NF-κB pathway. J. Neurochem. 2019, 151, 595–607. [Google Scholar] [CrossRef]
- Ritter, B.; Greten, F.R. Modulating inflammation for cancer therapy. J. Exp. Med. 2019, 216, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V.; Moinuddin. AGEs, RAGEs and s-RAGE; friend or foe for cancer. Semin. Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Mo, F.; Chang, B.; Zhang, Q.; Ran, H.; Yang, S.; Zhu, Z.; Hu, L.; Su, Q. Glucose-derived AGEs enhance human gastric cancer metastasis through RAGE/ERK/Sp1/MMP2 cascade. Oncotarget 2017, 8, 104216–104226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolonin, M.G.; Sergeeva, A.; Staquicini, D.I.; Smith, T.L.; Tarleton, C.A.; Molldrem, J.J.; Sidman, R.L.; Marchiò, S.; Pasqualini, R.; Arap, W. Interaction between Tumor Cell Surface Receptor RAGE and Proteinase 3 Mediates Prostate Cancer Metastasis to Bone. Cancer Res. 2017, 77, 3144–3150. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Chen, K.C.; Chang, G.C.; Lin, H.; Wu, C.C.; Kao, W.H.; Teng, C.L.J.; Hsu, S.L.; Yang, T.Y. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Miyazaki, A.; Ikemoto, M.; Arai, H.; Horiuchi, S.; Nakayama, H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J. Biol. Chem. 2001, 276, 13348–13355. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [Green Version]
- Elangovan, I.; Thirugnanam, S.; Chen, A.; Zheng, G.; Bosland, M.C.; Kajdacsy-Balla, A.; Gnanasekar, M. Targeting receptor for advanced glycation end products (RAGE) expression induces apoptosis and inhibits prostate tumor growth. Biochem. Biophys. Res. Commun. 2012, 417, 1133–1138. [Google Scholar] [CrossRef]
- Jandial, R.; Neman, J.; Lim, P.P.; Tamae, D.; Kowolik, C.M.; Wuenschell, G.E.; Shuck, S.C.; Ciminera, A.K.; De Jesus, L.R.; Ouyang, C.; et al. Inhibition of GLO1 in glioblastoma multiforme increases DNA-AGEs, stimulates RAGE expression, and inhibits brain tumor growth in orthotopic mouse models. Int. J. Mol. Sci. 2018, 19, 406. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, L.; Zhang, I.Y.; Liang, J.B. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res. 2014, 74, 7285–7297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef]
- Chen, M.; Glenn, J.V.; Dasari, S.; McVicar, C.; Ward, M.; Colhoun, L.; Quinn, M.; Bierhaus, A.; Xu, H.; Stitt, A.W. RAGE regulates immune cell infiltration and angiogenesis in choroidal neovascularization. PLoS ONE 2014, 9, e89548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Sotokawauchi, A.; Fukami, K.; Higashimoto, Y.; Yamagishi, S.I. RAGE-aptamer Attenuates the Growth and Liver Metastasis of Malignant Melanoma in Nude Mice. Mol. Med. 2017, 23, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Gianì, F.; Vella, V.; Tumino, D.; Malandrino, P.; Frasca, F. The possible role of cancer stem cells in the resistance to kinase inhibitors of advanced thyroid cancer. Cancers 2020, 12, 2249. [Google Scholar] [CrossRef]
- Takano, T. Fetal cell carcinogenesis of the thyroid: A modified theory based on recent evidence. Endocr. J. 2014, 61, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerashchenko, T.S.; Denisov, E.V.; Litviakov, N.V.; Zavyalova, M.V.; Vtorushin, S.V.; Tsyganov, M.M.; Perelmuter, V.M.; Cherdyntseva, N.V. Intratumor heterogeneity: Nature and biological significance. Biochemistry 2013, 78, 1201–1215. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Figueroa, H.; Morales, E. Fueling inflammation at tumor microenvironment: The role of multiligand/rage axis. Carcinogenesis 2010, 31, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Coluccio, M.L.; Presta, I.; Greco, M.; Gervasi, R.; La Torre, D.; Renne, M.; Voci, C.P.; Lunelli, L.; Donato, G.; Malara, N. Microenvironment molecular profile combining glycation adducts and cytokines patterns on secretome of short-term blood-derived cultures during tumour progression. Int. J. Mol. Sci. 2020, 21, 4711. [Google Scholar] [CrossRef]
- Zheng, J.; Lu, W.; Wang, C.; Xing, Y.; Chen, X.; Ail, Z. Galectin-3 induced by hypoxia promotes cell migration in thyroid cancer cells. Oncotarget 2017, 8, 101475–101488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Chiavarina, B.; Nokin, M.J.; Bellier, J.; Durieux, F.; Bletard, N.; Sherer, F.; Lovinfosse, P.; Peulen, O.; Verset, L.; Dehon, R.; et al. Methylglyoxal-mediated stress correlates with high metabolic activity and promotes tumor growth in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondrogianni, N.; Gonos, E.S. Proteasome inhibition induces a senescence-like phenotype in primary human fibroblasts cultures. Biogerontology 2004, 5, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kuzan, A.; Królewicz, E.; Nowakowska, K.; Stach, K.; Kaliszewki, K.; Domasławski, P.; Kotyra, Ł.; Gamian, A.; Kustrzeba-Wójcicka, I. Contribution of glycation and oxidative stress to thyroid gland pathology—A pilot study. Biomolecules 2021, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Włodarczyk, M.; Nowicka, G. Obesity, DNA damage, and development of obesity-related diseases. Int. J. Mol. Sci. 2019, 20, 1146. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Van Bezu, J.; Van Der Schors, R.C.; Uchida, K.; Stehouwer, C.D.A.; Van Hinsbergh, V.W.M. Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. FEBS Lett. 2006, 580, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatone, C.; Heizenrieder, T.; Di Emidio, G.; Treffon, P.; Amicarelli, F.; Seidel, T.; Eichenlaub-Ritter, U. Evidence that carbonyl stress by methylglyoxal exposure induces DNA damage and spindle aberrations, affects mitochondrial integrity in mammalian oocytes and contributes to oocyte ageing. Hum. Reprod. 2011, 26, 1843–1859. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.F.; Roque, L.; Krug, T.; Leite, V. Poorly differentiated and anaplastic thyroid carcinomas: Chromosomal and oligo-array profile of five new cell lines. Br. J. Cancer 2007, 96, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Russo, D.; Durante, C.; Bulotta, S.; Puppin, C.; Puxeddu, E.; Filetti, S.; Damante, G. Targeting histone deacetylase in thyroid cancer. Expert Opin. Ther. Targets 2013, 17, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, J.M.; Rabbani, G.; Ahmad, S.; Hasan, Q.; Khan, R.H.; Alam, K.; Choi, I.C. Glycation of H1 histone by 3-deoxyglucosone: Effects on protein structure and generation of different advanced glycation end products. PLoS ONE 2015, 10, e0130630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Omans, N.D.; Leicher, R.; Osunsade, A.; Agustinus, A.S.; Finkin-Groner, E.; D’Ambrosio, H.; Liu, B.; Chandarlapaty, S.; Liu, S.; et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 2019, 10, 1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular histones, cell-free DNA, or nucleosomes: Differences in immunostimulation. Cell Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef] [Green Version]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. Age-rage axis stimulates oxidized LDL uptake into macrophages through cyclin-dependent kinase 5-cd36 pathway via oxidative stress generation. Int. J. Mol. Sci. 2020, 21, 9263. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Iovino, F.; Eterno, V.; Cammareri, P.; Gambara, G.; Espina, V.; Gulotta, G.; Dieli, F.; Giordano, S.; De Maria, R.; et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010, 70, 8874–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, T.; Cuny, J.; Nawroth, G.; Djuric, Z.; Humpert, P.M.; Zeier, M.; Bierhaus, A.; Nawroth, P.P. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia 2012, 55, 1151–1155. [Google Scholar] [CrossRef] [Green Version]
- Bansode, S.B.; Chougale, A.D.; Joshi, R.S.; Giri, A.P.; Bodhankar, S.L.; Harsulkar, A.M.; Kulkarni, M.J. Proteomic analysis of protease resistant proteins in the diabetic rat kidney. Mol. Cell. Proteom. 2013, 12, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H2O2 and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef]
- Antognelli, C.; Mancuso, F.; Frosini, R.; Arato, I.; Calvitti, M.; Calafiore, R.; Talesa, V.N.; Luca, G. testosterone and follicle stimulating hormone–dependent glyoxalase 1 up-regulation sustains the viability of porcine sertoli cells through the control of hydroimidazolone– and argpyrimidine-mediated NF-κB pathway. Am. J. Pathol. 2018, 188, 2553–2563. [Google Scholar] [CrossRef] [Green Version]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, O.S.; Kim, C.S.; Sohn, E.; Jo, K.; Kim, J.S. Accumulation of argpyrimidine, a methylglyoxal-derived advanced glycation end product, increases apoptosis of lens epithelial cells both in vitro and in vivo. Exp. Mol. Med. 2012, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447. [Google Scholar] [CrossRef]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, C.; Nicolò, A.; Prevenzano, I.; Formisano, P.; Beguinot, F.; Miele, C. The dual-role of methylglyoxal in tumor progression—Novel therapeutic approaches. Front. Oncol. 2021, 11, 645686. [Google Scholar] [CrossRef]
- Lin, J.A.; Wu, C.H.; Yen, G.C. Methylglyoxal displays colorectal cancer-promoting properties in the murine models of azoxymethane and CT26 isografts. Free Radic. Biol. Med. 2018, 115, 436–446. [Google Scholar] [CrossRef]
- Antognelli, C.; Mandarano, M.; Prosperi, E.; Sidoni, A.; Talesa, V.N. Glyoxalase-1-Dependent Methylglyoxal Depletion Sustains PD-L1 Expression in Metastatic Prostate Cancer Cells: A novel mechanism in cancer immunosurveillance escape and a potential novel target to overcome PD-L1 blockade resistance. Cancers 2021, 13, 2965. [Google Scholar] [CrossRef]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal acts as a tumor-promoting factor in anaplastic thyroid cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.R.; Zafereo, M.E.; Dadu, R.; Ferrarotto, R.; Busaidy, N.L.; Lu, C.; Ahmed, S.; Gule-Monroe, M.K.; Williams, M.D.; Sturgis, E.M.; et al. Complete Surgical Resection Following Neoadjuvant Dabrafenib Plus Trametinib in BRAFV600E-Mutated Anaplastic Thyroid Carcinoma. Thyroid 2019, 29, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Smolarczyk, R.; Cichoń, T.; Jarosz, M.; Szala, S. HMGB1—Its role in tumor progression and anticancer therapy. Postepy Hig. Med. Dosw. (Online) 2012, 66, 913–920. [Google Scholar] [CrossRef]
- Davies, E.J.; Durban, V.M.; Meniel, V.; Williams, G.T.; Clarke, A.R. PTEN loss and KRAS activation leads to the formation of serrated adenomas and metastatic carcinoma in the mouse intestine. J. Pathol. 2014, 233, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS mutations in thyroid cancer. Oncologist 2013, 18, 926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poghosyan, Z.; Wynford-Thomas, D. Analysis of Ras Transformation of human thyroid epithelial cells. Methods Enzymol. 2006, 407, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, M.T.; Liu, A.; Xu, X.; Master, S.R.; Baldwin, D.A.; Tobias, J.W.; Livolsi, V.A.; Baloch, Z.W. Differential expression of miRNAs in papillary thyroid carcinoma compared to multinodular goiter using formalin fixed paraffin embedded tissues. Endocr. Pathol. 2007, 18, 163–173. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shirvani-Farsani, Z.; Taheri, M. The role of microRNAs in the pathogenesis of thyroid cancer. Non-Coding RNA Res. 2020, 5, 88. [Google Scholar] [CrossRef]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M.; et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef] [Green Version]
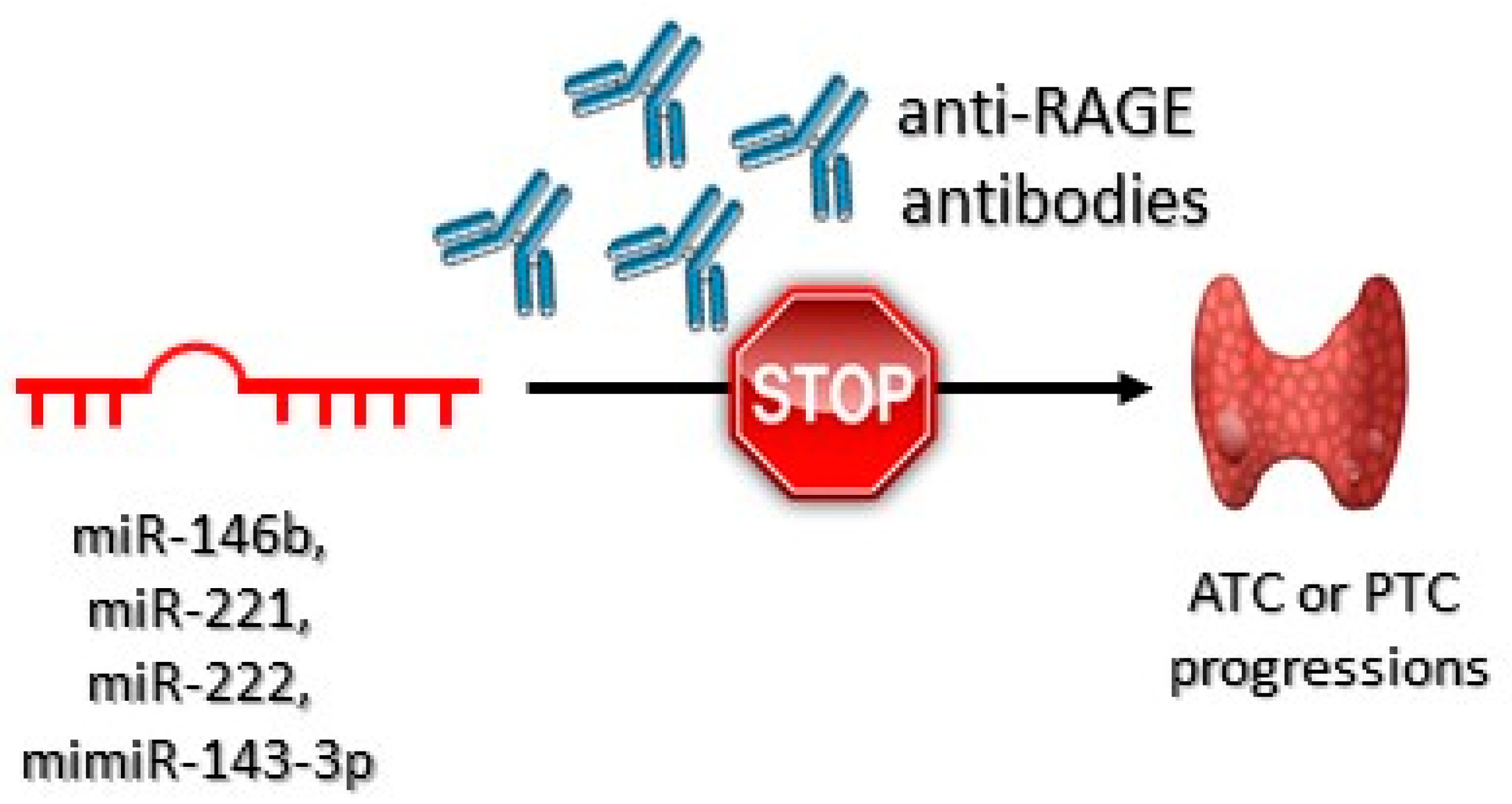
- Mardente, S.; Mari, E.; Consorti, F.; Di Gioia, C.; Negri, R.; Etna, M.; Zicari, A.; Antonaci, A. HMGB1 induces the overexpression of miR-222 and miR-221 and increases growth and motility in papillary thyroid cancer cells. Oncol. Rep. 2012, 28, 2285–2289. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Human miR-221/222 in physiological and atherosclerotic vascular Remodeling. BioMed Res. Int. 2015, 2015, 354517. [Google Scholar] [CrossRef]
- Helfman, D.M.; Kim, E.J.; Lukanidin, E.; Grigorian, M. The metastasis associated protein S100A4: Role in tumour progression and metastasis. Br. J. Cancer 2005, 92, 1955–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [Green Version]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE mediates the pro-migratory response of extracellular S100A4 in human thyroid cancer cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Arai, K.; Nozawa, R.; Yoshida, H.; Hirokawa, M.; Fukushima, M.; Inoue, H.; Tomoda, C.; Kihara, M.; Higashiyama, T.; et al. S100A8 and S100A9 expression is a crucial factor for dedifferentiation in thyroid carcinoma. Anticancer Res. 2009, 29, 4157–4161. [Google Scholar] [PubMed]
- Reeb, A.N.; Li, W.; Sewell, W.; Marlow, L.A.; Tun, H.W.; Smallridge, R.C.; Copland, J.A.; Spradling, K.; Chernock, R.; Lin, R.Y. S100A8 is a novel therapeutic target for anaplastic thyroid carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.S.; Hamdy, F.C.; Deloulme, J.C.; Rehman, I. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 2005, 46, 256–269. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bronowicka-Szydełko, A.; Kotyra, Ł.; Lewandowski, Ł.; Gamian, A.; Kustrzeba-Wójcicka, I. Role of Advanced Glycation End-Products and Other Ligands for AGE Receptors in Thyroid Cancer Progression. J. Clin. Med. 2021, 10, 4084. https://doi.org/10.3390/jcm10184084
Bronowicka-Szydełko A, Kotyra Ł, Lewandowski Ł, Gamian A, Kustrzeba-Wójcicka I. Role of Advanced Glycation End-Products and Other Ligands for AGE Receptors in Thyroid Cancer Progression. Journal of Clinical Medicine. 2021; 10(18):4084. https://doi.org/10.3390/jcm10184084
Chicago/Turabian StyleBronowicka-Szydełko, Agnieszka, Łukasz Kotyra, Łukasz Lewandowski, Andrzej Gamian, and Irena Kustrzeba-Wójcicka. 2021. "Role of Advanced Glycation End-Products and Other Ligands for AGE Receptors in Thyroid Cancer Progression" Journal of Clinical Medicine 10, no. 18: 4084. https://doi.org/10.3390/jcm10184084
APA StyleBronowicka-Szydełko, A., Kotyra, Ł., Lewandowski, Ł., Gamian, A., & Kustrzeba-Wójcicka, I. (2021). Role of Advanced Glycation End-Products and Other Ligands for AGE Receptors in Thyroid Cancer Progression. Journal of Clinical Medicine, 10(18), 4084. https://doi.org/10.3390/jcm10184084