
Exploring the Diverse Immune and Genetic Landscape of Psoriatic Arthritis


Abstract
:1. Introduction: Burden of Psoriatic Arthritis and Importance of Early Diagnosis
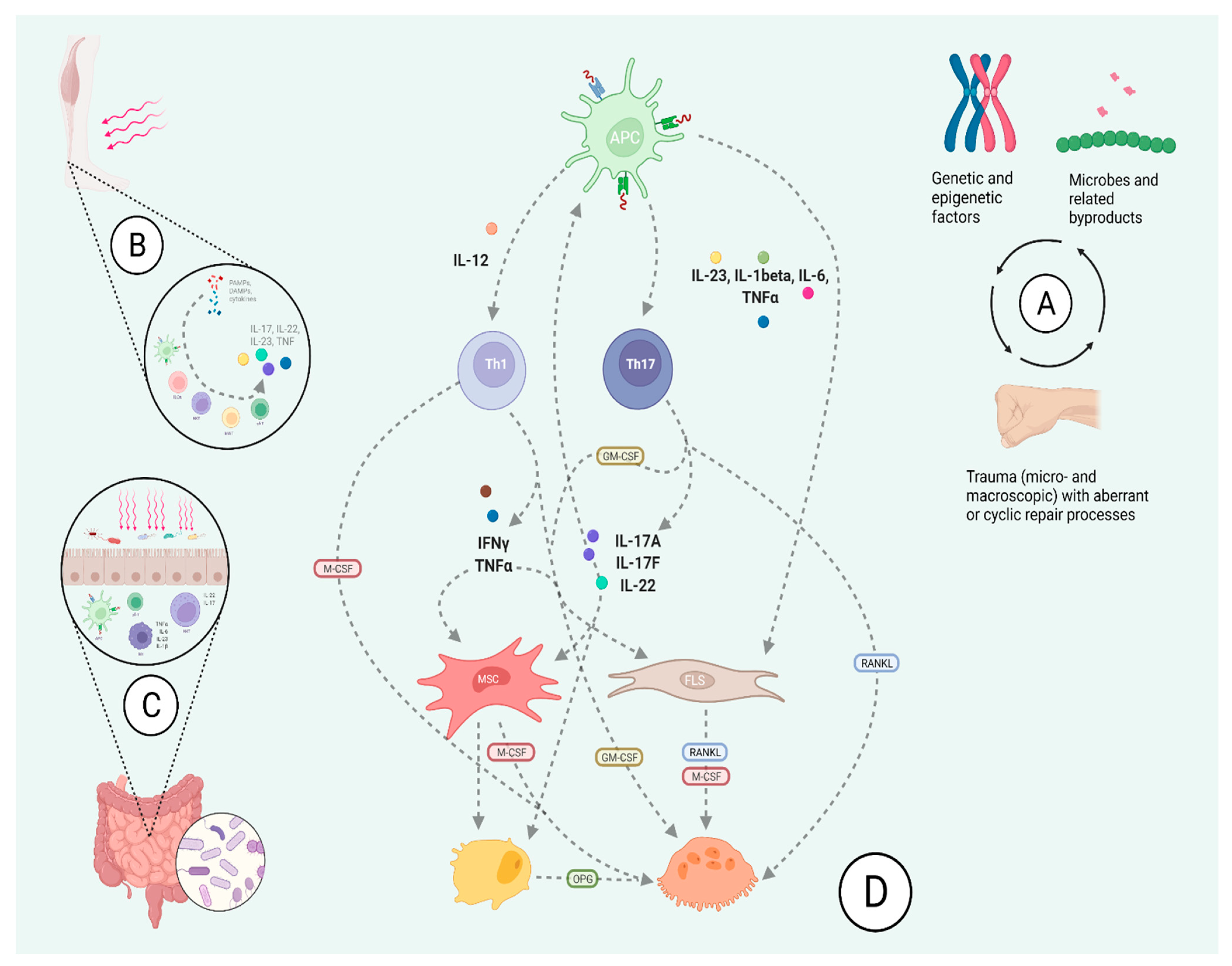
2. Wide Scope: A Broad Overview on the Pathobiology of Psoriatic Arthritis
3. Genetic Profile May Shape Disease Phenotype
4. The IL-23-Th17 Axis Shapes the Molecular Landscape of Skin and Joint Pathology in Psoriatic Arthritis
4.1. Moving beyond Singular Cytokine Effects—Synergistic Relationship between TNF Alpha and IL-17
4.2. IL-23 Signaling Leads to Psoriatic Skin and Joint Disease
5. Inflammation Is Co-Morbidity, but Not all Inflammation Is the Same
6. Summary
Funding
Conflicts of Interest
References
- Kane, D.; Stafford, L.; Bresnihan, B.; FitzGerald, O. A prospective, clinical and radiological study of early psoriatic arthritis: An early synovitis clinic experience. Rheumatology 2003, 42, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Christophers, E.; Barker, J.; Griffiths, C.; Daudén, E.; Milligan, G.; Molta, C.; Sato, R.; Boggs, R. The risk of psoriatic arthritis remains constant following initial diagnosis of psoriasis among patients seen in European dermatology clinics. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Batko, B. Patient-Centered Care in Psoriatic Arthritis—A Perspective on Inflammation, Disease Activity, and Psychosocial Factors. J. Clin. Med. 2020, 9, 3103. [Google Scholar] [CrossRef] [PubMed]
- Batko, B.; Kucharz, E.; Stajszczyk, M.; Brzosko, M.; Samborski, W.; Żuber, Z. Real-World Data from a Multi-Center Study: Insights to Psoriatic Arthritis Care. J. Clin. Med. 2021, 10, 4106. [Google Scholar] [CrossRef] [PubMed]
- Orbai, A.M.; Reddy, S.M.; Dennis, N.; Villacorta, R.; Peterson, S.; Mesana, L.; Chakravarty, S.D.; Lin, I.; Karyekar, C.S.; Wang, Y.; et al. Work absenteeism and disability associated with psoriasis and psoriatic arthritis in the USA—A retrospective study of claims data from 2009 to 2020. Clin. Rheumatol. 2021, 40, 4933–4942. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.C.; Icen, M.; Crowson, C.S.; McEvoy, M.T.; Gabriel, S.E.; Kremers, H.M. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: A population-based study. Arthritis Rheum. 2009, 61, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and incidence of psoriatic arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Thavaneswaran, A.; Chandran, V.; Cook, R.J. Do patients with psoriatic arthritis who present early fare better than those presenting later in the disease? Ann. Rheum. Dis. 2011, 70, 2152–2154. [Google Scholar] [CrossRef] [PubMed]
- Haroon, M.; Gallagher, P.; FitzGerald, O. Diagnostic delay of more than 6 months contributes to poor radiographic and functional outcome in psoriatic arthritis. Ann. Rheum. Dis. 2014, 74, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Felbo, S.; Terslev, L.; Sørensen, I.; Skov, L.; Zachariae, C.; Østergaard, M. Musculoskeletal Pain in Patients with Psoriasis and its Influence on Health-related Quality of Life: Results from a Danish Population-based Survey. Acta Derm. Venereol. 2021, 101, adv00553. [Google Scholar] [CrossRef] [PubMed]
- Ingrasciotta, Y.; Isgrò, V.; Ientile, V.; Tari, M.; Trifirò, G.; Guarneri, C. Are Patients with Psoriasis and Psoriatic Arthritis Undertreated? A Population-Based Study from Southern Italy. J. Clin. Med. 2021, 10, 3431. [Google Scholar] [CrossRef]
- Dolcino, M.; Lunardi, C.; Ottria, A.; Tinazzi, E.; Patuzzo, G.; Puccetti, A. Crossreactive Autoantibodies Directed against Cutaneous and Joint Antigens Are Present in Psoriatic Arthritis. PLoS ONE 2014, 9, e115424. [Google Scholar] [CrossRef]
- Belasco, J.; Louie, J.S.; Gulati, N.; Wei, N.; Nograles, K.; Fuentes-Duculan, J.; Mitsui, H.; Suárez-Fariñas, M.; Krueger, J.G. Comparative Genomic Profiling of Synovium Versus Skin Lesions in Psoriatic Arthritis. Arthritis Rheumatol. 2014, 67, 934–944. [Google Scholar] [CrossRef]
- Kurilenko, N.; Fatkhullina, A.; Mazitova, A.; Koltsova, E. Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis. Cells 2021, 10, 348. [Google Scholar] [CrossRef]
- Marchini, T.; Hansen, S.; Wolf, D. ApoB-Specific CD4+ T Cells in Mouse and Human Atherosclerosis. Cells 2021, 10, 446. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-F.; Chen, Y.-H.; Liu, S.-F.; Kao, H.-L.; Wu, M.-S.; Yang, K.-C.; Wu, W.-K. Mutual Interplay of Host Immune System and Gut Microbiota in the Immunopathology of Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 8729. [Google Scholar] [CrossRef]
- Łukasik, Z.; Gracey, E.; Venken, K.; Ritchlin, C.; Elewaut, D. Crossing the boundaries: IL-23 and its role in linking inflammation of the skin, gut and joints. Rheumatology 2021, 60, iv16–iv27. [Google Scholar] [CrossRef]
- Olejniczak-Staruch, I.; Ciążyńska, M.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. Alterations of the Skin and Gut Microbiome in Psoriasis and Psoriatic Arthritis. Int. J. Mol. Sci. 2021, 22, 3998. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Bierkarre, H.; Harder, J.; Cuthbert, R.; Emery, P.; Leuschner, I.; Mrowietz, U.; Hedderich, J.; McGonagle, D.; Gläser, R. Differential expression of antimicrobial peptides in psoriasis and psoriatic arthritis as a novel contributory mechanism for skin and joint disease heterogeneity. Scand. J. Rheumatol. 2015, 45, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wu, X.; Rocha, C.S.; Rolston, M.; Garcia-Melchor, E.; Huynh, M.; Nguyen, M.; Law, T.; Haas, K.N.; Yamada, D.; et al. Short-Term Western Diet Intake Promotes IL-23–Mediated Skin and Joint Inflammation Accompanied by Changes to the Gut Microbiota in Mice. J. Investig. Dermatol. 2021, 141, 1780–1791. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. Suppression of gut dysbiosis reverses Western diet-induced vascular dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E468–E477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadon, D.R.; Stober, C.; Pennington, S.R.; FitzGerald, O. Applying precision medicine to unmet clinical needs in psoriatic disease. Nat. Rev. Rheumatol. 2020, 16, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Stober, C. Pathogenesis of psoriatic arthritis. Best Pr. Res. Clin. Rheumatol. 2021, 35, 101694. [Google Scholar] [CrossRef] [PubMed]
- Eder, L.; Law, T.; Chandran, V.; Shanmugarajah, S.; Shen, H.; Rosen, C.F.; Cook, R.J.; Gladman, D.D. Association between environmental factors and onset of psoriatic arthritis in patients with psoriasis. Arthritis Rheum. 2011, 63, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Pattison, E.; Harrison, B.J.; Griffiths, C.E.M.; Silman, A.J.; Bruce, I.N. Environmental risk factors for the development of psoriatic arthritis: Results from a case-control study. Ann. Rheum. Dis. 2007, 67, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, O.B.; Svendsen, A.J.; Ejstrup, L.; Skytthe, A.; Junker, P. On the heritability of psoriatic arthritis. Disease concordance among monozygotic and dizygotic twins. Ann. Rheum. Dis. 2008, 67, 1417–1421. [Google Scholar] [CrossRef] [Green Version]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. Am. J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef] [Green Version]
- Vasey, F.B.; Deitz, C.; A Fenske, N.; Germain, B.F.; Espinoza, L.R. Possible involvement of group A streptococci in the pathogenesis of psoriatic arthritis. J. Rheumatol. 1982, 9. [Google Scholar]
- Filer, C.; Ho, P.; Smith, R.L.; Griffiths, C.; Young, H.; Worthington, J.; Bruce, I.N.; Barton, A. Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis Rheum. 2008, 58, 3705–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowes, J.; Ashcroft, J.; Dand, N.; Jalali-Najafabadi, F.; Bellou, E.; Ho, P.; Marzo-Ortega, H.; Helliwell, P.S.; Feletar, M.; Ryan, A.; et al. Cross-phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann. Rheum. Dis. 2017, 76, 1774–1779. [Google Scholar] [CrossRef]
- Gladman, D.D.; Shuckett, R.; Russell, M.L.; Thorne, J.C.; Schachter, R.K. Psoriatic Arthritis (PSA)—An Analysis of 220 Patients. QJM Int. J. Med. 1987, 62, 127–141. [Google Scholar] [CrossRef]
- Pollock, R.A.; Abji, F.; Liang, K.; Chandran, V.; Pellett, F.J.; Virtanen, C.; Gladman, D.D. Gene Expression Differences between Psoriasis Patients with and without Inflammatory Arthritis. J. Investig. Dermatol. 2015, 135, 620–623. [Google Scholar] [CrossRef] [Green Version]
- Apel, M.; Uebe, S.; Bowes, J.; Giardina, E.; Korendowych, E.; Juneblad, K.; Pasutto, F.; Ekici, A.B.; McManus, R.; Ho, P.; et al. Variants in RUNX3 Contribute to Susceptibility to Psoriatic Arthritis, Exhibiting Further Common Ground with Ankylosing Spondylitis. Arthritis Rheum. 2013, 65, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Eder, L.; Chandran, V.; Pellet, F.; Shanmugarajah, S.; Rosen, C.F.; Bull, S.B.; Gladman, D.D. Human leucocyte antigen risk alleles for psoriatic arthritis among patients with psoriasis. Ann. Rheum. Dis. 2011, 71, 50–55. [Google Scholar] [CrossRef]
- Haroon, M.; Winchester, R.; Giles, J.T.; Heffernan, E.; Fitzgerald, O. Clinical and genetic associations of radiographic sacroiliitis and its different patterns in psoriatic arthritis. Clin. Exp. Rheumatol. 2016, 35, 270–276. [Google Scholar] [PubMed]
- Haroon, M.; Winchester, R.; Giles, J.T.; Heffernan, E.; FitzGerald, O. Certain class I HLA alleles and haplotypes implicated in susceptibility play a role in determining specific features of the psoriatic arthritis phenotype. Ann. Rheum. Dis. 2014, 75, 155–162. [Google Scholar] [CrossRef]
- Kruithof, E.; Baeten, D.; De Rycke, L.; Vandooren, B.; Foell, D.; Roth, J.; Cañete, J.D.; Boots, A.M.; Veys, E.M.; De Keyser, F. Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res. 2005, 7, R569–R580. [Google Scholar] [CrossRef] [Green Version]
- Winchester, R.; Minevich, G.; Steshenko, V.; Kirby, B.; Kane, D.; Greenberg, D.A.; Fitzgerald, O. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2011, 64, 1134–1144. [Google Scholar] [CrossRef]
- Winchester, R.; FitzGerald, O. The many faces of psoriatic arthritis: Their genetic determinism. Rheumatology 2020, 59, i4–i9. [Google Scholar] [CrossRef] [Green Version]
- Dand, N.; Duckworth, M.; Baudry, D.; Russell, A.; Curtis, C.; Lee, S.H.; Evans, I.; Mason, K.; Alsharqi, A.; Becher, G.; et al. HLA-C*06:02 genotype is a predictive biomarker of biologic treatment response in psoriasis. J. Allergy Clin. Immunol. 2018, 143, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Van Vugt, L.J.; van den Reek, J.M.P.; Hannink, G.; Coenen, M.; De Jong, E.M.G.J. Association of HLA-C*06:02 Status with Differential Response to Ustekinumab in Patients with Psoriasis: A Systematic Review and Meta-Analysis. JAMA Dermatol. 2019, 155, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Q.; Yang, Z.; Wang, W.; Li, B.; Bai, M.; Wu, J.; Ge, H.; Dong, Z.; Shen, J.; Tang, H.; et al. Genetic Study on Small Insertions and Deletions in Psoriasis Reveals a Role in Complex Human Diseases. J. Investig. Dermatol. 2019, 139, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C.M.; Ramírez, C.P.; Martín, A.S.; Maroun, S.V.; Santiago, S.A.; Tortosa, M.R.; Morales, A.J. Influence of Genetic Polymorphisms on Response to Biologics in Moderate-to-Severe Psoriasis. J. Pers. Med. 2021, 11, 293. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Hedrich, C.M. The Molecular Pathophysiology of Psoriatic Arthritis—The Complex Interplay between Genetic Predisposition, Epigenetics Factors, and the Microbiome. Front. Mol. Biosci. 2021, 8, 662047. [Google Scholar] [CrossRef]
- Mulder, M.L.M.; He, X.; Reek, J.M.P.A.V.D.; Urbano, P.C.M.; Kaffa, C.; Wang, X.; van Cranenbroek, B.; van Rijssen, E.; Hoogen, F.H.J.V.D.; Joosten, I.; et al. Blood-Based Immune Profiling Combined with Machine Learning Discriminates Psoriatic Arthritis from Psoriasis Patients. Int. J. Mol. Sci. 2021, 22, 10990. [Google Scholar] [CrossRef]
- Gill, T.; Rosenbaum, J.T. Putative Pathobionts in HLA-B27-Associated Spondyloarthropathy. Front. Immunol. 2021, 11, 3510. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; MacDuff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [PubMed]
- Harris, T.J.; Grosso, J.F.; Yen, H.-R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting Edge: An in vivo Requirement for STAT3 Signaling in TH17 Development and TH17-Dependent Autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef] [Green Version]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010, 62, 2876–2885. [Google Scholar] [CrossRef]
- Raychaudhuri, S.P.; Raychaudhuri, S.K.; Genovese, M.C. IL-17 receptor and its functional significance in psoriatic arthritis. Mol. Cell. Biochem. 2011, 359, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, S.; Canavan, M.; McGarry, T.; Low, C.; Wade, S.C.; Mullan, R.H.; Veale, D.J.; Fearon, U. Association of synovial tissue polyfunctional T-cells with DAPSA in psoriatic arthritis. Ann. Rheum. Dis. 2019, 78, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Baricza, E.; Marton, N.; Királyhidi, P.; Kovacs, O.T.; Székely, I.K.; Lajkó, E.; Kőhidai, L.; Rojkovich, B.; Érsek, B.; Buzás, E.I.; et al. Distinct in vitro T-Helper 17 Differentiation Capacity of Peripheral Naive T Cells in Rheumatoid and Psoriatic Arthritis. Front. Immunol. 2018, 9, 606. [Google Scholar] [CrossRef] [Green Version]
- Dolcino, M.; Ottria, A.; Barbieri, A.; Patuzzo, G.; Tinazzi, E.; Argentino, G.; Beri, R.; Lunardi, C.; Puccetti, A. Gene Expression Profiling in Peripheral Blood Cells and Synovial Membranes of Patients with Psoriatic Arthritis. PLoS ONE 2015, 10, e0128262. [Google Scholar] [CrossRef] [Green Version]
- Noack, M.; Ndongo-Thiam, N.; Miossec, P. Interaction among activated lymphocytes and mesenchymal cells through podoplanin is critical for a high IL-17 secretion. Arthritis Res. 2016, 18, 148. [Google Scholar] [CrossRef] [Green Version]
- Celis, R.; Planell, N.; Fernández-Sueiro, J.L.; Sanmartí, R.; Ramírez, J.; González-Álvaro, I.; Pablos, J.L.; Cañete, J.D. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res. Ther. 2012, 14, R93. [Google Scholar] [CrossRef] [Green Version]
- Nerviani, A.; Boutet, M.-A.; Tan, W.S.G.; Goldmann, K.; Purkayastha, N.; Lajtos, T.A.; Hands, R.; Lewis, M.; Kelly, S.; Pitzalis, C. IL-23 skin and joint profiling in psoriatic arthritis: Novel perspectives in understanding clinical responses to IL-23 inhibitors. Ann. Rheum. Dis. 2020, 80, 591–597. [Google Scholar] [CrossRef]
- König, A.; Krenn, V.; Gillitzer, R.; Glöckner, J.; Jansen, E.; Gohlke, F.; Eulert, J.; Muller-Hermelink, H.K. Inflammatory infiltrate and interleukin-8 expression in the synovium of psoriatic arthritis—An immunohistochemical and mRNA analysis. Rheumatol. Int. 1997, 17, 159–168. [Google Scholar] [CrossRef]
- Menon, B.; Gullick, N.J.; Walter, G.J.; Rajasekhar, M.; Garrood, T.; Evans, H.G.; Taams, L.S.; Kirkham, B.W. Interleukin-17+CD8+ T Cells are Enriched in the Joints of Patients with Psoriatic Arthritis and Correlate with Disease Activity and Joint Damage Progression. Arthritis Rheumatol. 2014, 66, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Steel, K.J.A.; Srenathan, U.; Ridley, M.; Durham, L.E.; Wu, S.; Ryan, S.; Hughes, C.D.; Chan, E.; Kirkham, B.W.; Taams, L.S. Polyfunctional, Proinflammatory, Tissue-Resident Memory Phenotype and Function of Synovial Interleukin-17A+ CD8+ T Cells in Psoriatic Arthritis. Arthritis Rheumatol. 2019, 72, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Leijten, E.F.; van Kempen, T.S.; Nordkamp, M.A.O.; Pouw, J.N.; Kleinrensink, N.J.; Vincken, N.L.; Mertens, J.; Balak, D.M.W.; Verhagen, F.H.; Hartgring, S.A.; et al. Tissue-Resident Memory CD8+ T Cells from Skin Differentiate Psoriatic Arthritis from Psoriasis. Arthritis Rheumatol. 2021, 73, 1220–1232. [Google Scholar] [CrossRef]
- Van Raemdonck, K.; Umar, S.; Palasiewicz, K.; Romay, B.; Volkov, S.; Arami, S.; Sweiss, N.; Shahrara, S. TLR7 endogenous ligands remodel glycolytic macrophages and trigger skin-to-joint crosstalk in psoriatic arthritis. Eur. J. Immunol. 2020, 51, 714–720. [Google Scholar] [CrossRef]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, M.; Koc, A.; Luszczykiewicz, G.; Ksiezopolska-Pietrzak, K.; Klimczak, E.; Chwalinska-Sadowska, H.; Maśliński, W. High Levels of IL-17 in Rheumatoid Arthritis Patients: IL-15 Triggers In Vitro IL-17 Production Via Cyclosporin A-Sensitive Mechanism. J. Immunol. 2000, 164, 2832–2838. [Google Scholar] [CrossRef] [Green Version]
- Infante-Duarte, C.; Horton, H.F.; Byrne, M.C.; Kamradt, T. Microbial Lipopeptides Induce the Production of IL-17 in Th Cells. J. Immunol. 2000, 165, 6107–6115. [Google Scholar] [CrossRef] [Green Version]
- Koenders, M.I.; Kolls, J.K.; Oppers-Walgreen, B.; van den Bersselaar, L.; Joosten, L.A.B.; Schurr, J.R.; Schwarzenberger, P.; van den Berg, W.B.; Lubberts, E. Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis Rheum. 2005, 52, 3239–3247. [Google Scholar] [CrossRef]
- Lubberts, E.; Joosten, L.A.B.; Oppers, B.; van den Bersselaar, L.; Coenen-de Roo, C.J.J.; Kolls, J.K.; Schwarzenberger, P.; van de Loo, F.A.J.; van den Berg, W.B. IL-1-Independent Role of IL-17 in Synovial Inflammation and Joint Destruction during Collagen-Induced Arthritis. J. Immunol. 2001, 167, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubberts, E.; Schwarzenberger, P.; Huang, W.; Schurr, J.R.; Peschon, J.J.; van den Berg, W.B.; Kolls, J.K. Requirement of IL-17 Receptor Signaling in Radiation-Resistant Cells in the Joint for Full Progression of Destructive Synovitis. J. Immunol. 2005, 175, 3360–3368. [Google Scholar] [CrossRef] [Green Version]
- Nakae, S.; Saijo, S.; Horai, R.; Sudo, K.; Mori, S.; Iwakura, Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl. Acad. Sci. USA 2003, 100, 5986–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, S.; Nambu, A.; Sudo, K.; Iwakura, Y. Suppression of Immune Induction of Collagen-Induced Arthritis in IL-17-Deficient Mice. J. Immunol. 2003, 171, 6173–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plater-Zyberk, C.; Joosten, L.A.B.; A Helsen, M.M.; I Koenders, M.; A Baeuerle, P.; van den Berg, W.B. Combined blockade of granulocyte-macrophage colony stimulating factor and interleukin 17 pathways potently suppresses chronic destructive arthritis in a tumour necrosis factor α-independent mouse model. Ann. Rheum. Dis. 2008, 68, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Fossiez, F.; Taupin, J.L.; Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998, 161, 409–414. [Google Scholar] [PubMed]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, D.V.; A Di Battista, J.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar]
- Lavocat, F.; Ndongo-Thiam, N.; Miossec, P. Interleukin-25 Produced by Synoviocytes Has Anti-inflammatory Effects by Acting as a Receptor Antagonist for Interleukin-17A Function. Front. Immunol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.-C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.C.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17–producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2007, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Van Baarsen, L.G.; Lebre, M.C.; Van Der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: Possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res. Ther. 2014, 16, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladman, D.D.; Orbai, A.-M.; Klitz, U.; Wei, J.C.-C.; Gallo, G.; Birt, J.; Rathmann, S.; Shrom, D.; Marzo-Ortega, H. Ixekizumab and complete resolution of enthesitis and dactylitis: Integrated analysis of two phase 3 randomized trials in psoriatic arthritis. Arthritis Res. 2019, 21, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaçi, D.; Blauvelt, A.; Reich, K.; Tsai, T.-F.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Hugot, S.; You, R.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J. Am. Acad. Dermatol. 2015, 73, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Glatt, S.; Baeten, D.; Baker, T.; Griffiths, M.; Ionescu, L.; Lawson, A.D.G.; Maroof, A.; Oliver, R.; Popa, S.; Strimenopoulou, F.; et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: Evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann. Rheum. Dis. 2017, 77, 523–532. [Google Scholar] [CrossRef]
- Uluçkan, Ö.; Jimenez, M.; Karbach, S.; Jeschke, A.; Graña, O.; Keller, J.; Busse, B.; Croxford, A.L.; Finzel, S.; Koenders, M.; et al. Chronic skin inflammation leads to bone loss by IL-17–mediated inhibition of Wnt signaling in osteoblasts. Sci. Transl. Med. 2016, 8, 330ra37. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fanok, M.H.; Mediero-Munoz, A.; Fogli, L.K.; Corciulo, C.; Abdollahi, S.; Cronstein, B.N.; Scher, J.U.; Koralov, S.B. Augmented Th17 Differentiation Leads to Cutaneous and Synovio-Entheseal Inflammation in a Novel Model of Psoriatic Arthritis. Arthritis Rheumatol. 2018, 70, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Maroof, A.; Gikas, P.; Mittal, G.; Keen, R.; Baeten, D.; Shaw, S.; Roberts, S.J. Dual neutralisation of IL-17F and IL-17A with bimekizumab blocks inflammation-driven osteogenic differentiation of human periosteal cells. RMD Open 2020, 6, e001306. [Google Scholar] [CrossRef]
- Saczonek, A.O.; Krajewska-Włodarczyk, M.; Kasprowicz-Furmańczyk, M.; Placek, W. Immunological Memory of Psoriatic Lesions. Int. J. Mol. Sci. 2020, 21, 625. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Ehst, B.; Wang, Z.; Leitenberger, J.; McClanahan, D.; De La Torre, R.; Sawka, E.; Ortega-Loayza, A.G.; Strunck, J.; Greiling, T.; Simpson, E.; et al. Synergistic induction of IL-23 by TNFα, IL-17A, and EGF in keratinocytes. Cytokine 2020, 138, 155357. [Google Scholar] [CrossRef]
- Shen, F.; Ruddy, M.J.; Plamondon, P.; Gaffen, S.L. Cytokines link osteoblasts and inflammation: Microarray analysis of interleukin-17- and TNF-α-induced genes in bone cells. J. Leukoc. Biol. 2004, 77, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Chabaud, M.; Page, G.; Miossec, P. Enhancing Effect of IL-1, IL-17, and TNF-α on Macrophage Inflammatory Protein-3α Production in Rheumatoid Arthritis: Regulation by Soluble Receptors and Th2 Cytokines. J. Immunol. 2001, 167, 6015–6020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beringer, A.; Thiam, N.; Molle, J.; Bartosch, B.; Miossec, P. Synergistic effect of interleukin-17 and tumour necrosis factor-α on inflammatory response in hepatocytes through interleukin-6-dependent and independent pathways. Clin. Exp. Immunol. 2018, 193, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaba, L.C.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte Overexpression of IL-17C Promotes Psoriasiform Skin Inflammation. J. Immunol. 2013, 190, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Najm, A.; McInnes, I.B. IL-23 orchestrating immune cell activation in arthritis. Rheumatology 2021, 60, iv4–iv15. [Google Scholar] [CrossRef]
- Melis, L.; Vandooren, B.; Kruithof, E.; Jacques, P.; De Vos, M.; Mielants, H.; Verbruggen, G.; De Keyser, F.; Elewaut, D. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann. Rheum. Dis. 2009, 69, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Durell, B.G.; Noelle, R.J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Investig. 2002, 110, 493–497. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Fiocco, U.; Accordi, B.; Martini, V.; Oliviero, F.; Facco, M.; Cabrelle, A.; Piva, L.; Molena, B.; Caso, F.; Costa, L.; et al. JAK/STAT/PKCδ molecular pathways in synovial fluid T lymphocytes reflect the in vivo T helper-17 expansion in psoriatic arthritis. Immunol. Res. 2014, 58, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogli, L.K.; Sundrud, M.S.; Goel, S.; Bajwa, S.; Jensen, K.; Derudder, E.; Sun, A.; Coffre, M.; Uyttenhove, C.; Van Snick, J.; et al. T Cell–Derived IL-17 Mediates Epithelial Changes in the Airway and Drives Pulmonary Neutrophilia. J. Immunol. 2013, 191, 3100–3111. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Capon, F.; Di Meglio, P.; Szaub, J.; Prescott, N.; Dunster, C.; Baumber, L.; Timms, K.; Gutin, A.; Abkevic, V.; Burden, A.D.; et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum. Genet. 2007, 122, 201–206. [Google Scholar] [CrossRef]
- Cargill, M.; Schrodi, S.; Chang, M.; Garcia, V.E.; Brandon, R.; Callis, K.P.; Matsunami, N.; Ardlie, K.G.; Civello, D.; Catanese, J.J.; et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. Am. J. Hum. Genet. 2007, 80, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.; et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.-J. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Nikamo, P.; Lysell, J.; Ståhle, M. Association with Genetic Variants in the IL-23 and NF-κB Pathways Discriminates between Mild and Severe Psoriasis Skin Disease. J. Investig. Dermatol. 2015, 135, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, H.L.; Kagami, S.; Phillips, K.G.; Kurtz, S.E.; Jacques, S.L.; Blauvelt, A. IL-23–Mediated Psoriasis-Like Epidermal Hyperplasia Is Dependent on IL-17A. J. Immunol. 2010, 186, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonel, G.; Conrad, C.; Laggner, U.; Di Meglio, P.; Grys, K.; McClanahan, T.K.; Blumenschein, W.M.; Qin, J.-Z.; Xin, H.; Oldham, E.; et al. Cutting Edge: A Critical Functional Role for IL-23 in Psoriasis. J. Immunol. 2010, 185, 5688–5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewood, C.; Watad, A.; Russell, T.; Palmer, T.M.; Marzo-Ortega, H.; Khan, A.; A Millner, P.; Dunsmuir, R.; Rao, A.; Loughenbury, P.; et al. Identification of myeloid cells in the human enthesis as the main source of local IL-23 production. Ann. Rheum. Dis. 2019, 78, 929–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Veldhoen, M.; Martin, B. Th17 T cells: Linking innate and adaptive immunity. Semin. Immunol. 2007, 19, 353–361. [Google Scholar] [CrossRef]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.J.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- Petermann, F.; Rothhammer, V.; Claussen, M.C.; Haas, J.D.; Blanco, L.R.; Heink, S.; Prinz, I.; Hemmer, B.; Kuchroo, V.K.; Oukka, M.; et al. γδ T Cells Enhance Autoimmunity by Restraining Regulatory T Cell Responses via an Interleukin-23-Dependent Mechanism. Immunity 2010, 33, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.K.; Abria, C.; Mitra, A.; Raychaudhuri, S.P. Functional significance of MAIT cells in psoriatic arthritis. Cytokine 2019, 125, 154855. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.; Murray, J.; Simpson, C.; Okoye, R.; Tyson, K.; Griffiths, M.; Baeten, D.; Shaw, S.; Maroof, A. Interleukin (IL)-12 and IL-18 Synergize to Promote MAIT Cell IL-17A and IL-17F Production Independently of IL-23 Signaling. Front. Immunol. 2020, 11, 585134. [Google Scholar] [CrossRef]
- Diani, M.; Casciano, F.; Marongiu, L.; Longhi, M.; Altomare, A.; Pigatto, P.D.; Secchiero, P.; Gambari, R.; Banfi, G.; Manfredi, A.A.; et al. Increased frequency of activated CD8+ T cell effectors in patients with psoriatic arthritis. Sci. Rep. 2019, 9, 10870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penkava, F.; Velasco-Herrera, M.D.C.; Young, M.D.; Yager, N.; Nwosu, L.N.; Pratt, A.G.; Lara, A.L.; Guzzo, C.; Maroof, A.; Mamanova, L.; et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat. Commun. 2020, 11, 4727. [Google Scholar] [CrossRef] [PubMed]
- Wierzbowska-Drabik, K.; Lesiak, A.; Skibińska, M.; Niedźwiedź, M.; Kasprzak, J.D.; Narbutt, J. Psoriasis and Atherosclerosis—Skin, Joints, and Cardiovascular Story of Two Plaques in Relation to the Treatment with Biologics. Int. J. Mol. Sci. 2021, 22, 10402. [Google Scholar] [CrossRef]
- Eid, R.E.; Rao, D.A.; Zhou, J.; Lo, S.-F.L.; Ranjbaran, H.; Gallo, A.; Sokol, S.I.; Pfau, S.; Pober, J.S.; Tellides, G. Interleukin-17 and Interferon-γ Are Produced Concomitantly by Human Coronary Artery–Infiltrating T Cells and Act Synergistically on Vascular Smooth Muscle Cells. Circulation 2009, 119, 1424–1432. [Google Scholar] [CrossRef] [Green Version]
- Hot, A.; Lenief, V.; Miossec, P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann. Rheum. Dis. 2012, 71, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Thorp, E.; Tabas, I. Identification of a Non-Growth Factor Role for GM-CSF in Advanced Atherosclerosis: Promotion of Macrophage Apoptosis and Plaque Necrosis through IL-23 Signaling. Circ. Res. 2015, 116, e13–e24. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, U.; Ali, R.; Lebastchi, A.H.; Qin, L.; Lo, S.-F.L.; Yakimov, A.O.; Khan, S.F.; Choy, J.; Geirsson, A.; Pober, J.S.; et al. IFN-γ Primes Intact Human Coronary Arteries and Cultured Coronary Smooth Muscle Cells to Double-Stranded RNA- and Self-RNA–Induced Inflammatory Responses by Upregulating TLR3 and Melanoma Differentiation-Associated Gene 5. J. Immunol. 2010, 185, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Yu, X.; Ding, Y.-J.; Fu, Q.-Q.; Xie, J.-J.; Tang, T.-T.; Yao, R.; Chen, Y.; Liao, Y.-H. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin. Immunol. 2008, 127, 89–97. [Google Scholar] [CrossRef]
- Xie, J.-J.; Wang, J.; Tang, T.-T.; Chen, J.; Gao, X.-L.; Yuan, J.; Zhou, Z.-H.; Liao, M.-Y.; Yao, R.; Yu, X.; et al. The Th17/Treg functional imbalance during atherogenesis in ApoE−/− mice. Cytokine 2010, 49, 185–193. [Google Scholar] [CrossRef]
- Batko, B.; Maga, P.; Urbanski, K.; Ryszawa-Mrozek, N.; Schramm-Luc, A.; Koziej, M.; Mikolajczyk, T.; McGinnigle, E.; Czesnikiewicz-Guzik, M.; Ceranowicz, P.; et al. Microvascular dysfunction in ankylosing spondylitis is associated with disease activity and is improved by anti-TNF treatment. Sci. Rep. 2018, 8, 13205. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Hollan, I.; Curran, S.A.; Kitson, S.M.; Riggio, M.P.; Mikkelsen, K.; Almdahl, S.M.; Aukrust, P.; McInnes, I.B.; Goodyear, C.S. Brief Report: Proatherogenic Cytokine Microenvironment in the Aortic Adventitia of Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Batko, B.; Schramm-Luc, A.; Skiba, D.S.; Mikolajczyk, T.P.; Siedlinski, M. TNF-α Inhibitors Decrease Classical CD14hiCD16− Monocyte Subsets in Highly Active, Conventional Treatment Refractory Rheumatoid Arthritis and Ankylosing Spondylitis. Int. J. Mol. Sci. 2019, 20, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberoi, R.; Schuett, J.; Schuett, H.; Koch, A.-K.; Luchtefeld, M.; Grote, K.; Schieffer, B. Targeting Tumor Necrosis Factor-α with Adalimumab: Effects on Endothelial Activation and Monocyte Adhesion. PLoS ONE 2016, 11, e0160145. [Google Scholar] [CrossRef]
- Oberoi, R.; Vlacil, A.-K.; Schuett, J.; Schösser, F.; Schuett, H.; Tietge, U.J.; Schieffer, B.; Grote, K. Anti-tumor necrosis factor-α therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis 2018, 277, 80–89. [Google Scholar] [CrossRef]
- Gabriel, A.S.; Martinsson, A.; Wretlind, B.; Ahnve, S. IL-6 levels in acute and post myocardial infarction: Their relation to CRP levels, infarction size, left ventricular systolic function, and heart failure. Eur. J. Intern. Med. 2004, 15, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.; Naime, D.; Ostad, E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Investig. 1993, 92, 2675–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alten, R.; Gómez-Reino, J.; Durez, P.; Beaulieu, A.; Sebba, A.; Krammer, G.; Preiss, R.; Arulmani, U.; Widmer, A.; Gitton, X.; et al. Efficacy and safety of the human anti-IL-1beta monoclonal antibody canakinumab in rheumatoid arthritis: Results of a 12-week, phase II, dose-finding study. BMC Musculoskelet. Disord. 2011, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- El-Zayadi, A.A.; Jones, E.A.; Churchman, S.M.; Baboolal, T.; Cuthbert, R.J.; El-Jawhari, J.; Badawy, A.M.; Alase, A.A.; El-Sherbiny, Y.; McGonagle, D. Interleukin-22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: A novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology 2016, 56, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Davelaar, N.; Mus, A.; Asmawidjaja, P.S.; Hazes, J.M.W.; Baeten, D.L.P.; Vis, M.; Bisoendial, R.J.; Prens, E.P.; Lubberts, E. Interleukin-17A Is Produced by CD4+ but Not CD8+ T Cells in Synovial Fluid Following T Cell Receptor Activation and Regulates Different Inflammatory Mediators Compared to Tumor Necrosis Factor in a Model of Psoriatic Arthritis Synovitis. Arthritis Rheumatol. 2020, 72, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Clinical Questions of Interest | Potential Justification |
|---|---|
| Are clinical manifestations in PsA consistent with the theorized point of origin? |
|
| Why is considerable heterogeneity present across spondyloarthritis and even psoriatic arthritis itself? |
|
| What can be responsible for variability in treatment response in PsA? | Clinical problem: Despite several biologic and small molecule drugs being extensively tested in PsA, the response rates remain suboptimal. Even drug changes with respect to cytokine-targets do not always alleviate refractory disease.
|
| Responses are based on the studies discussed in this review and particularly [1,17,23,24,32,40,48,57,90]. | |
| Reference | Design | Detailed Summary |
|---|---|---|
| Sherlock et al. [48] | Murine model | IL-23 inhibition reduces entheseal inflammation, which is associated with downregulation of cytokines (e.g., IL-6, IL-1beta), chemokines (Cxcl1 and Cxcl2) and factors involved in bone remodeling (Rankl, Ctsk, MMPs). In both axial and peripheral articular surfaces, IL-23R+ resident cells are present in entheses. This population is characterized by “innate-like” responsiveness, which may confer responsiveness to IL-23 in entheses, as seen in the gut.Early on enthesitis is present without synovitis, late in the disease, destructive synovitis and florid enthesitis are present. IL-23 leads to joint inflammation (early changes in entheses and periosteum) and follows a dose-related relationship, though no disease in other organs develops (kidney, liver, gut). IL-23 leads to enthesitis in axial skeleton and sacroiliitis. IL-23 leads to expansion of periosteal osteoblasts, and new entheseal and periosteal cartilage and bone formation. IL-23-driven models do not fully respond to TNF, IL-6 or RANKL inhibition.Th17 is not necessary for IL-23-related inflammatory disease to develop (but rather local IL-23R+ resident cells). IL-23 stimulates IL-17 and IL-22. Inhibition of IL-17 and IL-22 reduces joint swelling, more so in combination. Of note, IL-17 overexpression does not lead to pathology. IL-22 has osteoproliferative effects. Joint swelling with phosphorylation of STAT3 in bone is associated with IL-22. In comparison to IL-23, induction of genes regulating bone formation (Wnt, bone morphogenic proteins, alkaline phosphatase) is more pronounced for IL-22. |
| Zayyadi et al. [141] | In vitro experiment based on human tissue | Inflammatory stimulus (IFN-gamma and TNF) enhances IL-22 receptor expression in mesenchymal stem cells (MSCs). MSC proliferation and migration are enhanced by concerted activity of IL-22 and inflammatory stimuli. IL-22 upregulates osteogenic markers. When IFN-gamma, TNF and IL-22 act together, chondrogenic and adipogenic transcription factor expression remains largely unaltered, except for reduced elevation of pro-osteogenic RUNX2. IL-22 related osteogenesis is reduced in the presence of inflammatory stimulus (i.e., TNF and IFN-gamma). |
| Baarsen et al. [83] | In vitro experiment based on human tissue | IL-17A is significantly elevated in synovium of inflammatory arthritis patients, but there is high heterogeneity across individuals. Receptor expression for IL-17A, IL-17F and respective receptors is highly variable in inflammatory synovium. IL-17 producing cells are widespread in synovium, with the majority being CD3+ T cells. CD4+ and CD8+ as well as CD68+ and CD163+ macrophages may be sources of IL-17. |
| Benham et al. [54] | In vitro experiment based on human tissue | IL-17+ and IL-22+ CD4+ T cells are elevated in peripheral blood mononuclear cells (PBMCs) of both PsA and PsO subjects, as compared with healthy subjects. IL-17 production is enhanced in both PsA and PsO, but IL-22 secretion is greater by PBMCs from PsA subjects (despite similar frequencies of IL-22+ cells). In PsA, increased frequency of CD4+ IL-17+ cells, and reduced CD4+ IL-22+ T cells are observed in synovial fluid. CD4+ IL-17+ T cells in synovial fluid mesenchymal cells were elevated, while CD4+ IL-22+ T cells were reduced (as compared with blood). In synovial tissue, IL-17 is not uniform, while IL-22 expression is absent. |
| Wade et al. [57] | In vitro experiment based on human tissue | T-cell polyfunctionality with regard to cytokine expression is enhanced in synovial tissue and associated with disease activity, in contrast to monofunctional T cells. PDE4 inhibition leads to suppression of polyfunctional cells. |
| Menon et al. [64] | In vitro experiment based on human tissue | PsA joints, but not those of RA patients, have enhanced levels of IL-17+ CD4- (CD8+) and IL-17+CD4+T cells. T cell subsets are associated with disease activity and erosive disease status. IL-17+ CD3+ T cells (with increased frequency of CD3+CD4- T cells and CD3+ CD4+ T cells) are elevated in synovial fluid of PsA, as compared to peripheral blood. The majority of IL-17+CD4- T cells are CD8+ or CD161+ T subsets, and a small proportion expressed characteristic markers of MAIT cells or γ/δ cells. Significant differences are observed in cytokine expression of T cells in matched peripheral blood and synovium in PsA. Synovial fluid IL-17+ CD4- T cells, but not CD4+ counterparts, are positively correlated with active synovitis scores. Moreover, IL-22+ CD4- T cells show a similar association. IL-17+CD4- T cells are elevated in synovial fluid from PsA subjects and the proportion of IL-17+ is increased in CD4- and CD4+ T cell populations when erosive disease is present, but not in nonerosive cases. |
| Baricza et al. [58] | In vitro experiment based on human tissue | Naive CD4+CD45RO− T lymphocytes are shown to be predisposed to shift to Th17 and produce IL-17A and IL-22. Increased RoR γ expression is present in naïve T cells of PsA patients. Cytokine combinations result in specific changes of transcription factors and IL-17A and IL-22 production in PsA. Chemokine receptor patterns suggest naïve T cells are likely to be prematurely engaged in PsA. |
| Uluckan et al. [87] | Murine model | Increase in Th17 cells is concurrent with reduction in other T helper and regulatory cells (i.e., Th1, Th2 and Treg cells, which may prevent osteoclastogenesis). Osteoclast progenitor cells are likely to accumulate and RANKL may be enhanced due to augmentation of Th17 responses (In the experimental model of R26STAT3Cstopfl/fl CD4Cre mice). Conversely, osteoblasts are characterized by failure to develop. Neutralization of IL-17 or genetic ablation of IL-22 alleviates the psoriasis phenotype. Abrogation of IL-22 and IL-17 (Th17 cytokines) prevents osteopenia. |
| Xu et al. [142] | In vitro experiment based on human tissue | CD4+ T cells are the major population in PsA synovial fluid and blood (as opposed to CD8+T cells). CD4+ T cells, but not CD8+ T cells are sources of IL-17A in synovial fluid of PsA patients following TCR activation. Anti-17A activity leads to more pronounced inhibition of inflammatory cytokines (e.g., IL-6 and IL-1beta), while TNF-alpha inhibition leads to stronger reduction in MMPs. |
| Mulder et al. [46] | In vitro experiment based on human tissue | Based on blood-based immune profiling, a reduction in CD4+ and CD8+ memory T-cell subsets, Treg cells and CD196+ and CD197+ monocytes in concert with elevated levels of differentiated CD4+ memory T-cells expressing CCR6 and CCR4 discriminates PsA from PsO. Memory T cells and CCR6+ monocytes are likely to migrate to articular and entheseal tissue, which could explain the differences with PsO peripheral blood. The increase in CD196+ (CCR6) memory T cells is considered to reflect a compensatory proliferation stimulus in response to their efflux to inflamed tissue. CD197+ (CCR7) monocytes are reduced in circulation of PsA subjects, and this subset is strongly associated with disease activity. This may reflect the recruitment of these populations into inflamed tissue, which is also supported by studies that show CCR7 signaling is related to Th-17-driven bone loss. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batko, B. Exploring the Diverse Immune and Genetic Landscape of Psoriatic Arthritis. J. Clin. Med. 2021, 10, 5926. https://doi.org/10.3390/jcm10245926
Batko B. Exploring the Diverse Immune and Genetic Landscape of Psoriatic Arthritis. Journal of Clinical Medicine. 2021; 10(24):5926. https://doi.org/10.3390/jcm10245926
Chicago/Turabian StyleBatko, Bogdan. 2021. "Exploring the Diverse Immune and Genetic Landscape of Psoriatic Arthritis" Journal of Clinical Medicine 10, no. 24: 5926. https://doi.org/10.3390/jcm10245926
APA StyleBatko, B. (2021). Exploring the Diverse Immune and Genetic Landscape of Psoriatic Arthritis. Journal of Clinical Medicine, 10(24), 5926. https://doi.org/10.3390/jcm10245926