
Prostacyclin Analogues Inhibit Platelet Reactivity, Extracellular Vesicle Release and Thrombus Formation in Patients with Pulmonary Arterial Hypertension

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Methods
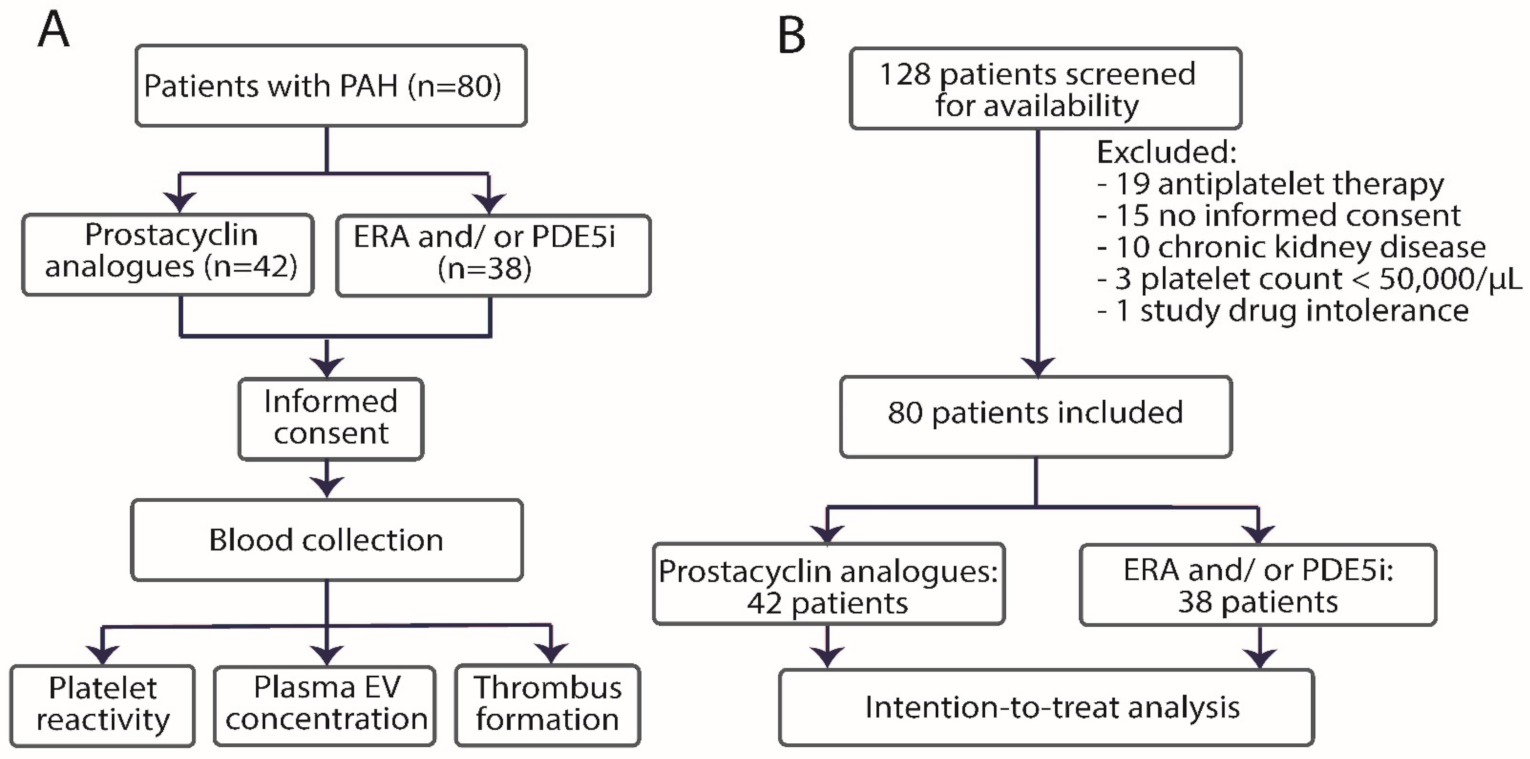
2.1. Study Design
2.2. Selection of Participants
2.3. Trial Schedule and Blinding
2.4. Study Treatment
2.5. Clinical Data Collection
2.6. Samples Collection and Handling
2.7. Endpoints
2.8. Laboratory Assays
2.8.1. Platelet Reactivity
2.8.2. Flow Cytometry
2.8.3. Whole Blood Perfusion System
2.9. Statistical Analysis
3. Results
3.1. Study Population
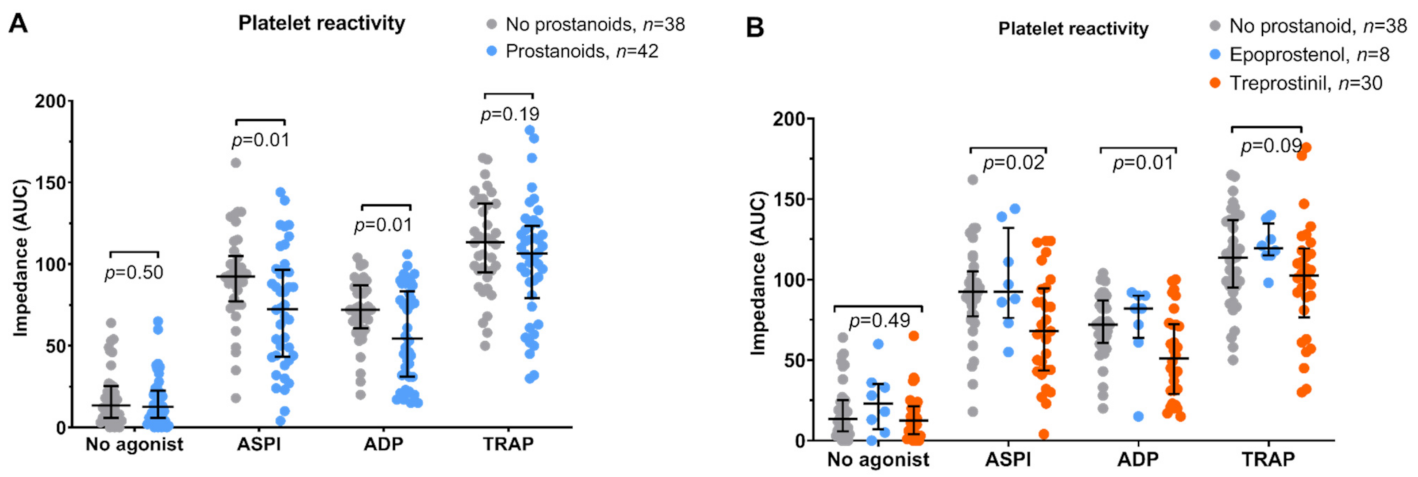
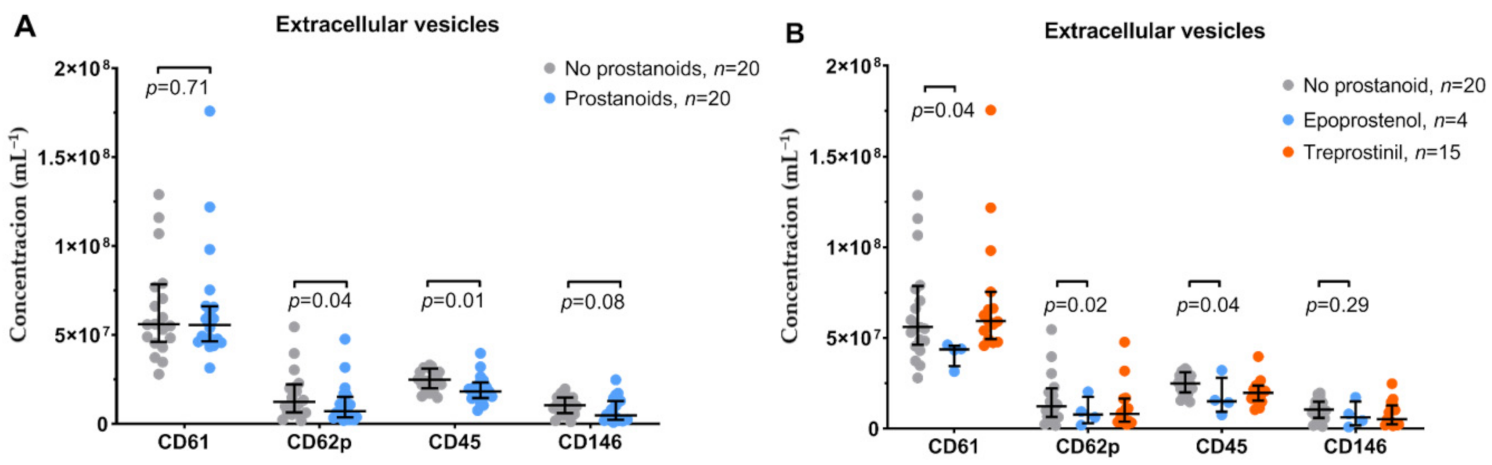
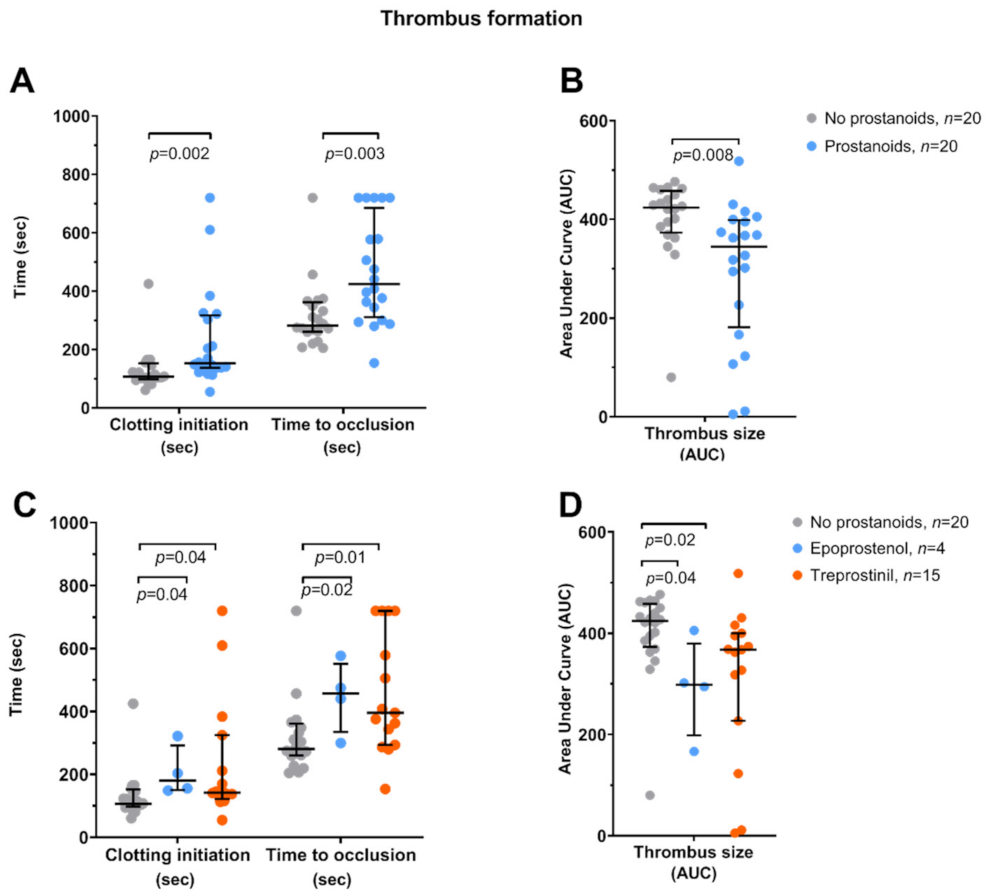
3.2. Platelet Function
3.3. Clinical Outcomes
3.4. Compliance
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of Disease: Pulmonary Arterial Hypertension. Nat. Rev. Cardiol. 2011, 8, 443. [Google Scholar] [CrossRef]
- Rabinovitch, M. Molecular Pathogenesis of Pulmonary Arterial Hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Phipps, R.P.; White, R.J. Thrombosis, Platelets, Microparticles and PAH: More than a Clot. Drug Discov. Today 2014, 19, 1230–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet Extracellular Vesicles as Biomarkers for Arterial Thrombosis. Platelets 2017, 28, 228–234. [Google Scholar] [CrossRef]
- Amabile, N.; Guignabert, C.; Montani, D.; Yeghiazarians, Y.; Boulanger, C.M.; Humbert, M. Cellular Microparticles in the Pathogenesis of Pulmonary Hypertension. Eur. Respir. J. 2013, 42, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Nadaud, S.; Poirier, O.; Girerd, B.; Blanc, C.; Montani, D.; Eyries, M.; Imbert-Bismut, F.; Pacheco, A.; Vigne, J.; Tregouet, D.-A.; et al. Small Platelet Microparticle Levels Are Increased in Pulmonary Arterial Hypertension. Eur. J. Clin. Investig. 2013, 43, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Matsubara, H. Increased Levels of Platelet-Derived Microparticles in Pulmonary Hypertension. Thromb. Res. 2020. [Google Scholar] [CrossRef]
- Muller, I.; Klocke, A.; Alex, M.; Kotzsch, M.; Luther, T.; Morgenstern, E.; Zieseniss, S.; Zahler, S.; Preissner, K.; Engelmann, B. Intravascular Tissue Factor Initiates Coagulation via Circulating Microvesicles and Platelets. FASEB J. 2003, 17, 476–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, L.A.; Haven, A.K.; Bauer, N.N. Circulating Microparticles in Severe Pulmonary Arterial Hypertension Increase Intercellular Adhesion Molecule-1 Expression Selectively in Pulmonary Artery Endothelium. Respir. Res. 2016, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cuesta, F.; Passalacqua, I.; Rodor, J.; Bhushan, R.; Denby, L.; Baker, A.H. Extracellular Vesicle Cross-Talk between Pulmonary Artery Smooth Muscle Cells and Endothelium during Excessive TGF-$β$ Signalling: Implications for PAH Vascular Remodelling. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amabile, N.; Heiss, C.; Real, W.M.; Minasi, P.; McGlothlin, D.; Rame, E.J.; Grossman, W.; De Marco, T.; Yeghiazarians, Y. Circulating Endothelial Microparticle Levels Predict Hemodynamic Severity of Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 1268–1275. [Google Scholar] [CrossRef]
- Amabile, N.; Heiss, C.; Chang, V.; Angeli, F.S.; Damon, L.; Rame, E.J.; McGlothlin, D.; Grossman, W.; De Marco, T.; Yeghiazarians, Y. Increased CD62e+ Endothelial Microparticle Levels Predict Poor Outcome in Pulmonary Hypertension Patients. J. Hear. lung Transplant. 2009, 28, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) Endors. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Kurzyna, M.; Araszkiewicz, A.; Błaszczak, P.; Grabka, M.; Hawranek, M.; Kopeć, G.; Mroczek, E.; Zembala, M.; Torbicki, A.; Ochała, A. Summary of Recommendations for the Haemodynamic and Angiographic Assessment of the Pulmonary Circulation. Joint Statement of the Polish Cardiac Society’s Working Group on Pulmonary Circulation and Association of Cardiovascular Interventions. Kardiol. Pol. Pol. Heart J. 2015, 73. [Google Scholar] [CrossRef] [Green Version]
- Kurzyna, M.; Małaczyńska-Rajpold, K.; Koteja, A.; Pawlak, A.; Chrzanowski, Ł.; Furdal, M.; Gąsior, Z.; Jacheć, W.; Sobkowicz, B.; Norwa, J.; et al. An Implantable Pump Lenus Pro®in the Treatment of Pulmonary Arterial Hypertension with Intravenous Treprostinil. BMC Pulm. Med. 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.J.; Wan, J.; Ghofrani, H.A.; Seeger, W.; Gall, H.; Rieth, A.; Tello, K. Acute Response to Rapid Iloprost Inhalation Using the BreelibTM Nebulizer in Pulmonary Arterial Hypertension: The BreelibTM Acute Study. Pulm. Circ. 2019, 9, 2045894019875342. [Google Scholar] [CrossRef] [Green Version]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.A.; Hill, A.F.A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Lacroix, R.; Robert, S.; Poncelet, P.; Kasthuri, R.S.; Key, N.S.; Dignat-George, F. Standardization of Platelet-Derived Microparticle Enumeration by Flow Cytometry with Calibrated Beads: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative Workshop. J. Thromb. Haemost. 2010, 8, 2571–2574. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Huczek, Z.; Kapłon-Cieślicka, A.; Lacroix, R.; Opolski, G.; et al. Randomized Controlled Trial Protocol to Investigate the Antiplatelet Therapy Effect on Extracellular Vesicles (AFFECT EV) in Acute Myocardial Infarction. Platelets 2020. [Google Scholar] [CrossRef]
- van der Pol, E.; de Rond, L.; Coumans, F.A.W.; Gool, E.L.; Böing, A.N.; Sturk, A.; Nieuwland, R.; van Leeuwen, T.G. Absolute Sizing and Label-Free Identification of Extracellular Vesicles by Flow Cytometry. Nanomed. Nanotechnol. Biol. Med. 2018, 14. [Google Scholar] [CrossRef]
- de Rond, L.; Libregts, S.F.W.M.; Rikkert, L.G.; Hau, C.M.; van der Pol, E.; Nieuwland, R.; van Leeuwen, T.G.; Coumans, F.A.W. Refractive Index to Evaluate Staining Specificity of Extracellular Vesicles by Flow Cytometry. J. Extracell. Vesicles 2019. [Google Scholar] [CrossRef] [Green Version]
- Welsh, J.A.; van der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A Framework for Standardized Reporting of Extracellular Vesicle Flow Cytometry Experiments. J. Extracell. Vesicles 2019. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Lacroix, R.; Leroyer, A.; Opolski, G.; Pluta, K.; et al. Ticagrelor Attenuates the Increase of Extracellular Vesicle Concentrations in Plasma after Acute Myocardial Infarction Compared to Clopidogrel. J. Thromb. Haemost. 2020, 18, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Al Ghaithi, R.; Mori, J.; Nagy, Z.; Maclachlan, A.; Hardy, L.; Philippou, H.; Hethershaw, E.; Morgan, N.V.; Senis, Y.A.; Harrison, P. Evaluation of the Total Thrombus-Formation System (T-TAS): Application to Human and Mouse Blood Analysis. Platelets 2019, 30, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Vrigkou, E.; Tsangaris, I.; Bonovas, S.; Kopterides, P.; Kyriakou, E.; Konstantonis, D.; Pappas, A.; Anthi, A.; Gialeraki, A.; Orfanos, S.E.; et al. Platelet and Coagulation Disorders in Newly Diagnosed Patients with Pulmonary Arterial Hypertension. Platelets 2019, 30, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Kevane, B.; Allen, S.; Walsh, K.; Egan, K.; Maguire, P.B.; Galligan, M.C.; Kenny, D.; Savage, R.; Doran, E.; Lennon, Á.; et al. Dual Endothelin-1 Receptor Antagonism Attenuates Platelet-Mediated Derangements of Blood Coagulation in Eisenmenger Syndrome. J. Thromb. Haemost. 2018, 16, 1572–1579. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, E.R.; Kapur, V.; Rodriguez, J.; Rastogi, S.; Rosanio, S. The Effects of Chronic Phosphodiesterase-5 Inhibitor Use on Different Organ Systems. Int. J. Impot. Res. 2007, 19, 139–148. [Google Scholar] [CrossRef]
- Müller, I.; Massberg, S.; Zierhut, W.; Binz, C.; Schuster, A.; Rüdiger-von Hoch, S.; Braun, S.; Gawaz, M. Effects of Aspirin and Clopidogrel versus Oral Anticoagulation on Platelet Function and on Coagulation in Patients with Nonvalvular Atrial Fibrillation (CLAFIB). Pathophysiol. Haemost. Thromb. 2002, 32, 16–24. [Google Scholar] [CrossRef]
- van der Pol, E.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R.; Coumans, F.A.W. Standardization of Extracellular Vesicle Measurements by Flow Cytometry through Vesicle Diameter Approximation. J. Thromb. Haemost. 2018, 16, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Weiss, H.J.; Turitto, V.T. Prostacyclin (Prostaglandin I2, PGI2) Inhibits Platelet Adhesion and Thrombus Formation on Subendothelium. Blood 1979, 53, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Higgs, E.A.; Higgs, G.A.; Moncada, S.; Vane, J.R. Prostacyclin (PGI2) Inhibits the Formation of Platelet Thrombi in Arterioles and Venules of the Hamster Cheek Pouch. Br. J. Pharmacol. 1978, 63, 535–539. [Google Scholar] [CrossRef]
- Yusuf, M.Z.; Raslan, Z.; Atkinson, L.; Aburima, A.; Thomas, S.G.; Naseem, K.M.; Calaminus, S.D.J. Prostacyclin Reverses Platelet Stress Fibre Formation Causing Platelet Aggregate Instability. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, S.; Küpper, J.-H.; Jung, F. Effect of Prostanoids on Human Platelet Function: An Overview. Int. J. Mol. Sci. 2020, 21, 9020. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Kirkby, N.S. Eicosanoids, Prostacyclin and Cyclooxygenase in the Cardiovascular System. Br. J. Pharmacol. 2019, 176, 1038–1050. [Google Scholar] [CrossRef]
- Van Heerden, P.V.; Gibbs, N.M.; Michalopoulos, N. Effect of Low Concentrations of Prostacyclin on Platelet Function in Vitro. Anaesth. Intensive Care 1997, 25, 343–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburrelli, C.; Crescente, M.; Izzi, B.; Barisciano, M.; Donati, M.B.; De Gaetano, G.; Cerletti, C. Epoprostenol Inhibits Human Platelet-Leukocyte Mixed Conjugate and Platelet Microparticle Formation in Whole Blood. Thromb. Res. 2011, 128, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Reber, G.; De Moerloose, P.; Vadas, L.; Chiappe, A.; Spahr-Schopfer, I.; Rimensberger, P.C. Aerosolized Iloprost Induces a Mild but Sustained Inhibition of Platelet Aggregation. Eur. Respir. J. 2002, 19, 518–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacha, N.; Levy, M.; Guerin, C.L.; le Bonniec, B.; Harroche, A.; Gaussem, P.; Israel-Biet, D.; Boulanger, C.M.; Smadja, D.M. Treprostinil Decreases Platelet Microparticles in Pediatric Pulmonary Hypertension. Circulation 2017, 136 (Suppl. 1), A18517. [Google Scholar]
- Bacha, N.C.; Levy, M.; Guerin, C.L.; Le Bonniec, B.; Harroche, A.; Szezepanski, I.; Renard, J.M.; Gaussem, P.; Israel-Biet, D.; Boulanger, C.M.; et al. Treprostinil Treatment Decreases Circulating Platelet Microvesicles and Their Procoagulant Activity in Pediatric Pulmonary Hypertension. Pediatr. Pulmonol. 2019, 54, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, A.; Matsubara, H.; Fujio, H.; Miyaji, K.; Nakamura, K.; Morita, H.; Saito, H.; Kusano, K.F.; Emori, T.; Date, H.; et al. Risk of Alveolar Hemorrhage in Patients with Primary Pulmonary Hypertension. Circ. J. 2005, 69, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, L.; Bair, N.; Banjac, S.; Dweik, R.A.; Tonelli, A.R. Subdural Hematomas in Pulmonary Arterial Hypertension Patients Treated with Protacyclin Analogs. Pulm. Circ. 2012, 2, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasecka, A.; Nieuwland, R.; van der Pol, E.; Hajji, N.; Ćwiek, A.; Pluta, K.; Konwerski, M.; Filipiak, K.J. P2Y12 Antagonist Ticagrelor Inhibits the Release of Procoagulant Extracellular Vesicles from Activated Platelets: Preliminary Results. Cardiol. J. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaniak, M.; Gkasecka, A.; Filipiak, K.J. Cell-Derived Microvesicles in Cardiovascular Diseases and Antiplatelet Therapy Monitoring—A Lesson for Future Trials? Current Evidence, Recent Progresses and Perspectives of Clinical Application. Int. J. Cardiol. 2017, 226, 93–102. [Google Scholar] [CrossRef]
- Armstrong, R.A. Platelet Prostanoid Receptors. Pharmacol. Ther. 1996, 72, 171–191. [Google Scholar] [CrossRef]
- Gasecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065. [Google Scholar] [CrossRef] [PubMed]
- Saniabadi, A.R.; Lowe, G.D.O.; Belch, J.J.F.; Barbenel, J.C.; Forbes, C.D. Effect of Prostacyclin (Epoprostenol) on the Aggregation of Human Platelets in Whole Blood in Vitro. Pathophysiol. Haemost. Thromb. 1984, 14, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Pydi, S.P.; Gleim, S.; Bhullar, R.P.; Hwa, J.; Dakshinamurti, S.; Chelikani, P. New Insights into Structural Determinants for Prostanoid Thromboxane A2 Receptor-and Prostacyclin Receptor-G Protein Coupling. Mol. Cell. Biol. 2013, 33, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmell, C.H.; Sefton, M.V.; Yeo, E.L. Platelet-Derived Microparticle Formation Involves Glycoprotein IIb-IIIa. Inhibition by RGDS and a Glanzmann’s Thrombasthenia Defect. J. Biol. Chem. 1993, 268, 14586–14589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
|
| Study Group (n = 42) | Control Group (n = 38) | ||||
|---|---|---|---|---|---|
| Parameter | Number (%) or Mean ± SD or Median (IQR) | Number (%) or Mean ± SD or Median (IQR) | p-Value | ||
| Age, years—mean ± SD | 49.5 | 15.9 | 55.5 | 15.7 | 0.11 |
| Female gender—number (%) | 34 | 81 | 28 | 74 | 0.27 |
| BMI—mean ± SD | 26.4 | 6.2 | 26.9 | 4.7 | 0.84 |
| PAH etiology—number (%) | |||||
| Congenital heart disease | 3 | 7 | 7 | 18 | 0.18 |
| Connective tissue disease | 3 | 7 | 6 | 16 | 0.29 |
| Idiopathic | 36 | 86 | 25 | 66 | 0.04 |
| WHO functional class—number (%) | |||||
| II | 18 | 43 | 21 | 55 | 0.38 |
| III | 21 | 50 | 15 | 40 | 0.38 |
| IV | 3 | 7 | 2 | 5 | 1.00 |
| Comorbidities—number (%) | |||||
| Arterial hypertension | 17 | 45 | 19 | 50 | 0.50 |
| Diabetes mellitus | 6 | 16 | 7 | 18 | 0.76 |
| Dyslipidemia | 10 | 26 | 9 | 24 | 1.00 |
| Smoking | 2 | 5 | 3 | 8 | 0.66 |
| History of CVD—number (%) | |||||
| Myocardial infarction | 3 | 8 | 5 | 13 | 0.47 |
| Stroke | 3 | 8 | 2 | 5 | 1.00 |
| Laboratory parameters | |||||
| ALT, U/L—median (IQR) | 17.5 | 12-21 | 19.5 | 12-27 | 0.28 |
| Creatinine, mg/dL—mean ± SD | 0.92 | 0.27 | 0.97 | 0.29 | 0.49 |
| Hemoglobin, g/dL—mean ± SD | 14.3 | 1.8 | 13.6 | 2.6 | 0.19 |
| Platelet count, *103/μL—mean ± SD | 189 | 48 | 196 | 55 | 0.63 |
| INR—median (IQR) | 1.08 | 1.04-1.17 | 1.09 | 0.99-1.31 | 0.68 |
| NTproBNP, pg/mL—median (IQR) | 563 | 125-1577 | 284 | 93-1498 | 0.59 |
| Pharmacotherapy—number (%) | |||||
| Prostacyclin analogue | 42 | 100 | 0 | 0 | n.a. |
| Endothelin receptor antagonist | 21 | 50 | 17 | 45 | 0.66 |
| Phosphodiesterase 5 inhibitor | 40 | 95 | 34 | 90 | 0.42 |
| Calcium channel blocker | 4 | 10 | 8 | 21 | 0.21 |
| Diuretic | 18 | 43 | 20 | 53 | 0.50 |
| β-blocker | 12 | 29 | 14 | 37 | 0.47 |
| Statin | 22 | 53 | 19 | 50 | 1.00 |
| Oral anticoagulant | 7 | 17 | 7 | 18 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsecka, A.; Banaszkiewicz, M.; Nieuwland, R.; van der Pol, E.; Hajji, N.; Mutwil, H.; Rogula, S.; Rutkowska, W.; Pluta, K.; Eyileten, C.; et al. Prostacyclin Analogues Inhibit Platelet Reactivity, Extracellular Vesicle Release and Thrombus Formation in Patients with Pulmonary Arterial Hypertension. J. Clin. Med. 2021, 10, 1024. https://doi.org/10.3390/jcm10051024
Gąsecka A, Banaszkiewicz M, Nieuwland R, van der Pol E, Hajji N, Mutwil H, Rogula S, Rutkowska W, Pluta K, Eyileten C, et al. Prostacyclin Analogues Inhibit Platelet Reactivity, Extracellular Vesicle Release and Thrombus Formation in Patients with Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2021; 10(5):1024. https://doi.org/10.3390/jcm10051024
Chicago/Turabian StyleGąsecka, Aleksandra, Marta Banaszkiewicz, Rienk Nieuwland, Edwin van der Pol, Najat Hajji, Hubert Mutwil, Sylwester Rogula, Wiktoria Rutkowska, Kinga Pluta, Ceren Eyileten, and et al. 2021. "Prostacyclin Analogues Inhibit Platelet Reactivity, Extracellular Vesicle Release and Thrombus Formation in Patients with Pulmonary Arterial Hypertension" Journal of Clinical Medicine 10, no. 5: 1024. https://doi.org/10.3390/jcm10051024
APA StyleGąsecka, A., Banaszkiewicz, M., Nieuwland, R., van der Pol, E., Hajji, N., Mutwil, H., Rogula, S., Rutkowska, W., Pluta, K., Eyileten, C., Postuła, M., Darocha, S., Huczek, Z., Opolski, G., Filipiak, K. J., Torbicki, A., & Kurzyna, M. (2021). Prostacyclin Analogues Inhibit Platelet Reactivity, Extracellular Vesicle Release and Thrombus Formation in Patients with Pulmonary Arterial Hypertension. Journal of Clinical Medicine, 10(5), 1024. https://doi.org/10.3390/jcm10051024