
Impact of Integrated Genetic Information on Diagnosis and Prognostication for Myeloproliferative Neoplasms in the Next-Generation Sequencing Era

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Molecular Analysis
2.3. Cytogenetic Study
2.4. Statistical Analyses
3. Results
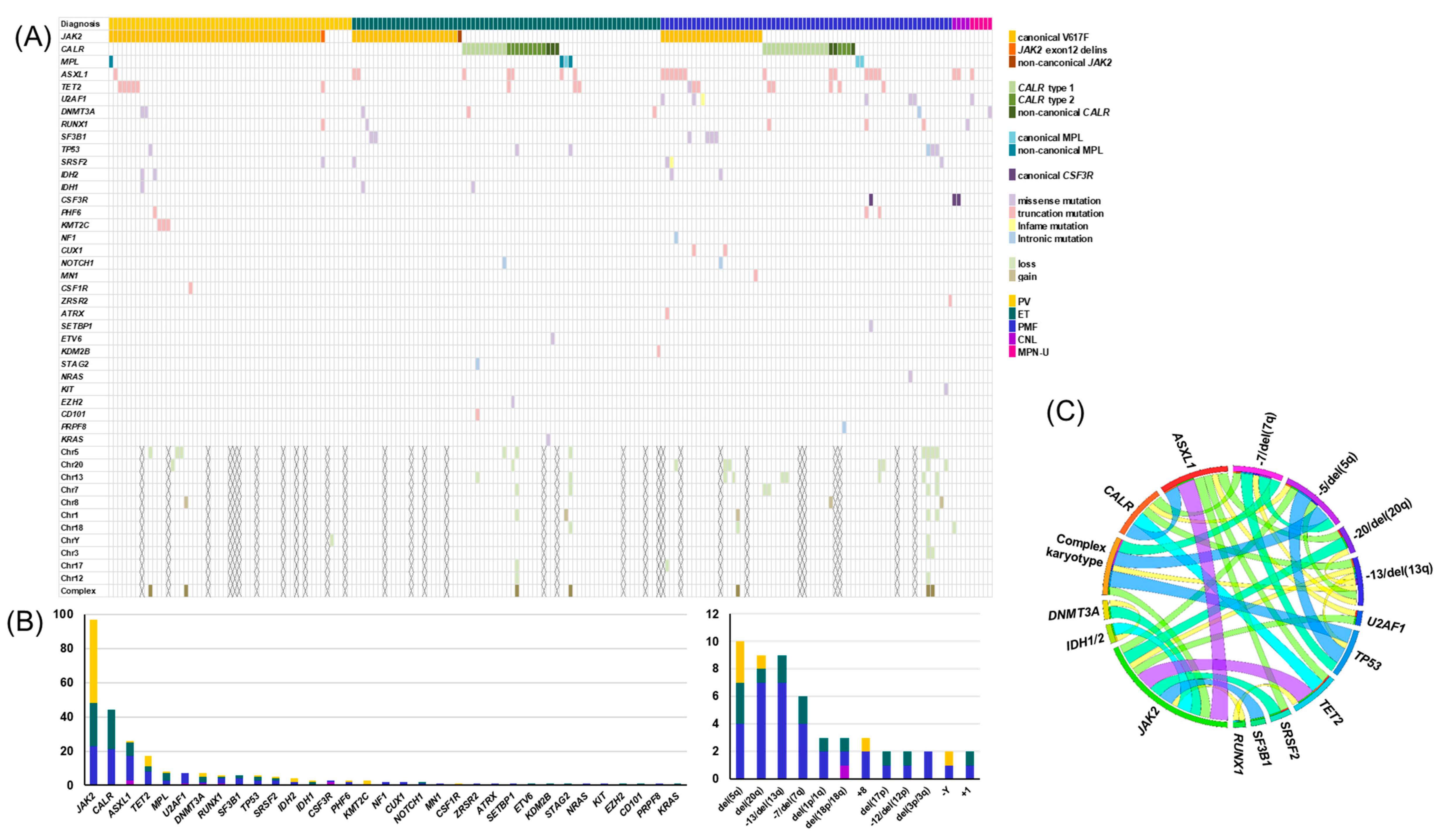
3.1. Genetic Landscape of MPNs
3.2. Impact of Genetic Aberrations on Diagnosis
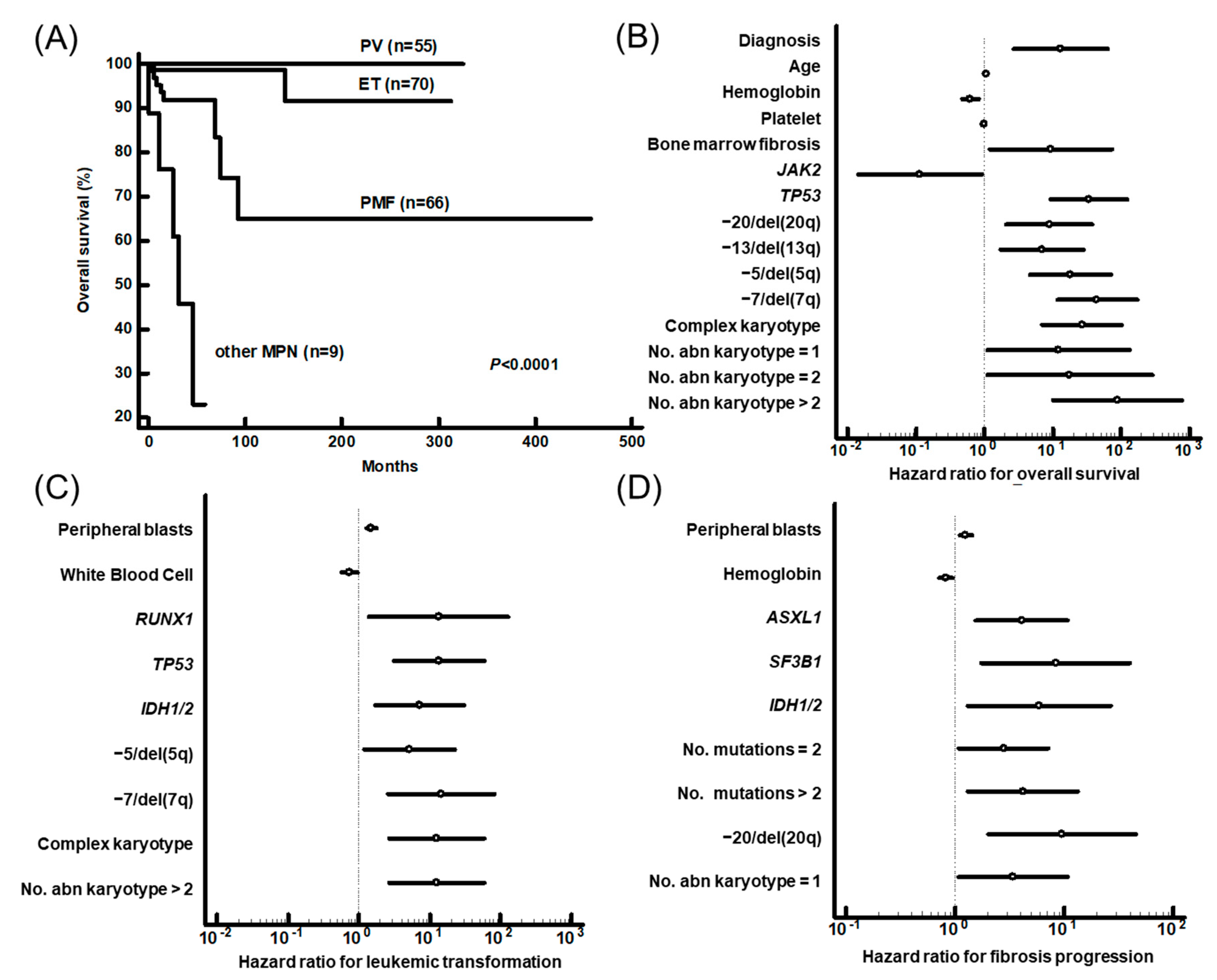
3.3. Prognostic Impact of Genetic Aberrations
3.4. Impact of Genetic Aberrations on Risk Stratification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; WHO: Geneva, Switzerland, 2017; Volume 2, pp. 30–59. [Google Scholar]
- Pardanani, A.; Lasho, T.; Laborde, R.; Elliott, M.; Hanson, C.; Knudson, R.; Ketterling, R.; Maxson, J.; Tyner, J.; Tefferi, A. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia 2013, 27, 1870–1873. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood J. Am. Soc. Hematol. 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martínez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood J. Am. Soc. Hematol. 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.; Finke, C.; Knudson, R.; Ketterling, R.; Hanson, C.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.-A.; Choi, C.W. Recent insights regarding the molecular basis of myeloproliferative neoplasms. Korean J. Intern. Med. 2020, 35, 1. [Google Scholar] [CrossRef] [Green Version]
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, C.J.; Panzarella, T.; Kennedy, J.A.; Arruda, A.; Claudio, J.O.; Daher-Reyes, G.; Ho, J.; Siddiq, N.; Devlin, R.; Tsui, H. The mutational landscape of accelerated-and blast-phase myeloproliferative neoplasms impacts patient outcomes. Blood Adv. 2018, 2, 2658–2671. [Google Scholar] [CrossRef] [Green Version]
- Kameda, T.; Shide, K.; Yamaji, T.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Marutsuka, K.; et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: Disease sustainer and disease accelerator. Blood 2015, 125, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Tefferi, A.; Levine, R.L. Role of TET2 and ASXL1 mutations in the pathogenesis of myeloproliferative neoplasms. Hematol. Oncol. Clin. 2012, 26, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.; Rodeghiero, F.; Randi, M.; Vaidya, R.; Cazzola, M.; Rambaldi, A. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Thiele, J.; Girodon, F.; Rumi, E.; Carobbio, A.; Gisslinger, H.; Kvasnicka, H.M.; Ruggeri, M.; Randi, M.L.; Gangat, N. A prognostic model to predict survival in 867 World Health Organization–defined essential thrombocythemia at diagnosis: A study by the International Working Group on Myelofibrosis Research and Treatment. Blood 2012, 120, 1197–1201. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood J. Am. Soc. Hematol. 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood J. Am. Soc. Hematol. 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V. MIPSS70: Mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F. Stem cell transplant in MF: It’s time to personalize. Blood J. Am. Soc. Hematol. 2019, 133, 2118–2120. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Thiele, J.; Ferrari, A.; Vannucchi, A.M.; Tefferi, A. The new WHO classification for essential thrombocythemia calls for revision of available evidences. Blood Cancer J. 2020, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Lee, J.-M.; Kim, Y.-J.; Park, S.-S.; Han, E.; Kim, M.; Kim, Y. Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome. J. Clin. Med. 2019, 8, 2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan-Jordan, J.; Simons, A.; Schmid, M. (Eds.) ISCN: An International System for Human Cytogenomic Nomenclature (2016); Karger: Basel, Switzerland, 2016. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1769. [Google Scholar] [CrossRef]
- Andréasson, B.; Pettersson, H.; Wasslavik, C.; Johansson, P.; Palmqvist, L.; Asp, J. ASXL1 mutations, previous vascular complications and age at diagnosis predict survival in 85 WHO-defined polycythaemia vera patients. Br. J. Haematol. 2020, 189, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Mutations and prognosis in myeloproliferative neoplasms. Leuk. Lymphoma 2019, 60, 1112–1113. [Google Scholar] [CrossRef]
- Jeromin, S.; Haferlach, T.; Grossmann, V.; Alpermann, T.; Kowarsch, A.; Haferlach, C.; Kern, W.; Schnittger, S. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica 2013, 98, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broséus, J.; Alpermann, T.; Wulfert, M.; Brichs, L.F.; Jeromin, S.; Lippert, E.; Rozman, M.; Lifermann, F.; Grossmann, V.; Haferlach, T. Age, JAK2 V617F and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2013, 27, 1826–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Lasho, T.L.; Gangat, N.; Begna, K.H.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Revised cytogenetic risk stratification in primary myelofibrosis: Analysis based on 1002 informative patients. Leukemia 2018, 32, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.; Kilpivaara, O.; Wadleigh, M. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic-or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E. JAK2/IDH-mutant–driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.; Lasho, T.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood J. Am. Soc. Hematol. 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematol. 2018, 183, 678–681. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Mañú, A.; Blasco-Iturri, Z.; Palomino-Echeverría, S.; Larrayoz, M.J.; García-Sanz, R.; Prieto-Conde, M.I.; del Carmen Chillón, M. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PLoS ONE 2020, 15, e0227986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izevbaye, I.; Liang, L.Y.; Mather, C.; El-Hallani, S.; Maglantay, R., Jr.; Saini, L. Clinical validation of a myeloid next-generation sequencing panel for single-nucleotide variants, Insertions/deletions, and fusion genes. J. Mol. Diagn. 2020, 22, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Kluk, M.J.; Lindsley, R.C.; Aster, J.C.; Lindeman, N.I.; Szeto, D.; Hall, D.; Kuo, F.C. Validation and implementation of a custom next-generation sequencing clinical assay for hematologic malignancies. J. Mol. Diagn. 2016, 18, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.; Smith, G.H.; Rossi, M.R.; Hill, C.E.; Zhang, L. Validation of a customized bioinformatics pipeline for a clinical next-generation sequencing test targeting solid tumor–associated variants. J. Mol. Diagn. 2018, 20, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Trosman, J.R.; Weldon, C.B.; Gradishar, W.J.; Benson, A.B., III; Cristofanilli, M.; Kurian, A.W.; Ford, J.M.; Balch, A.; Watkins, J.; Phillips, K.A. From the past to the present: Insurer coverage frameworks for next-generation tumor sequencing. Value Health 2018, 21, 1062–1068. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Variables | Total N = 200 | PV N = 55 | ET N = 70 | PMF N = 66 | Other MPN a N = 9 | p |
|---|---|---|---|---|---|---|
| Age at diagnosis, mean ± SD | 49.0 ± 14.7 | 50.3 ± 12.3 | 43.2 ± 15.3 | 52.5 ± 13.6 | 61.4 ± 14.2 | <0.001 |
| Sex, male (%) | 43.0 (86/200) | 52.7 (29/55) | 35.7 (25/70) | 39.4 (26/66) | 66.7 (6/9) | 0.109 |
| Follow-up months, mean ± SD | 64.7 ± 69.4 | 76.4 ± 67.7 | 78.5 ± 73.8 | 46.0 ± 64.8 | 23.6 ± 20.2 | <0.001 |
| White blood cells (109/l), mean ± SD | 12.9 ± 13.7 | 12.9 ± 7.4 | 9.5 ± 5.5 | 11.8 ± 11.9 | 47.8 ± 37.3 | <0.001 |
| Hemoglobin (g/l), mean ± SD | 129.3 ± 33.6 | 164.7 ± 31.3 | 126.2 ± 20.6 | 107.7 ± 21.3 | 95.3 ± 18.7 | <0.001 |
| Platelet (109/l), mean ± SD | 560.5 ± 484.8 | 542.2 ± 324.9 | 803.7 ± 608.7 | 367.5 ± 340.3 | 197.2 ± 147.5 | <0.001 |
| Bone marrow fibrosis, % | 46.0 (92/200) | 16.4 (9/55) | 21.4 (15/70) | 100.0 (66/66) | 22.2 (2/9) | 0.064 |
| Splenomegaly, % | 31.5 (63/200) | 27.3 (15/55) | 11.4 (8/70) | 54.5 (36/66) | 44.4 (4/9) | 0.364 |
| Vascular event b, % | 20.0 (40/200) | 36.4 (20/55) | 20.0 (14/70) | 9.1 (6/66) | 0 (0/9) | 0.275 |
| Abnormal karyotype c, % | 22.5 (38/169) | 18.6 (8/43) | 9.8 (6/61) | 37.5 (21/56) | 33.3 (3/9) | 0.003 |
| Complex karyotype c, % | 4.7 (8/169) | 4.7 (2/43) | 3.3 (2/61) | 7.1 (4/56) | 0 (0/9) | 0.692 |
| Number of mutations, mean ± SD | 1.3 ± 1.0 | 1.4 ± 0.9 | 1.1 ± 0.9 | 1.6 ± 1.1 | 0.9 ± 0.9 | 0.024 |
| PV | ET | PMF | 3 MPNs a | Other MPN b | |
|---|---|---|---|---|---|
| Case number | 55 | 70 | 66 | 191 | 9 |
| Triple mutations c | 49 (89.1%) | 51 (72.9%) | 46 (69.7%) | 146 (76.4%) | 2 (22.2%) |
| Any of the seven mutations d | 0 (0%) | 2 (2.9%) | 6 (9.1%) | 8 (4.2%) | 3 (33.3%) |
| Other mutations e and/or abnormal karyotypes | 1 (1.8%) | 1 (1.4%) | 9 (13.6%) | 11 (5.8%) | 4 (22.4%) |
| Any clonal genetic marker f | 50 (90.9%) | 54 (77.1%) | 61 (92.4%) | 165 (86.4%) | 7 (77.8%) |
| All negative | 5 (9.1%) | 16 (22.9%) | 5 (7.6%) | 26 (13.6%) | 2 (22.2%) |
| Variables | Overall Survival | Leukemic Transformation | Fibrotic Progression | ||||||
|---|---|---|---|---|---|---|---|---|---|
| P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | |
| Diagnosis a | 0.0087 | 78.2 | 3.0–2027.3 | ||||||
| PB blasts (%) | 0.0486 | 1.3 | 1.0-1.6 | ||||||
| No. mutation | 0.0352 | 2.0 | 1.1–4.0 | ||||||
| ASXL1 | 0.0358 | 4.3 | 1.1–16.4 | ||||||
| RUNX1 | 0.005 | 68.1 | 3.6–1300.4 | ||||||
| SF3B1 | 0.0009 | 31.5 | 4.1–243.3 | ||||||
| TP53 | 0.0041 | 64.2 | 3.8–1096.5 | 0.0364 | 16.3 | 1.2–222.7 | |||
| IDH1/2 | 0.0051 | 32.5 | 2.8–371.1 | 0.0011 | 21.2 | 3.4–132.2 | |||
| −7/del(7q) | 0.0219 | 14.0 | 1.5–132.7 | ||||||
| del(20q) | 0.0002 | 44.5 | 6.1–323.0 | ||||||
| Risk Group | Low | Intermediate | High | P (Contingency Coefficient) |
|---|---|---|---|---|
| MIPSS-PV | 18 | 23 | 14 | <0.001 (0.514) |
| Low | 18 | 14 | 6 | |
| Intermediate | 0 | 9 | 5 | |
| High | 0 | 0 | 3 | |
| MIPSS-ET | 42 | 24 | 4 | <0.001 (0.554) |
| Low | 41 | 19 | 0 | |
| Intermediate | 1 | 3 | 2 | |
| High | 0 | 2 | 2 |
| DIPSS | Low | Int-1 | Int-2 | High | P (Contingency Coefficient) |
|---|---|---|---|---|---|
| MIPSS70 | 25 | 16 | 21 | 4 | <0.001 (0.620) |
| Low | 15 | 1 | 0 | 0 | |
| Intermediate | 8 | 12 | 11 | 0 | |
| High | 2 | 3 | 10 | 4 | |
| MIPSS70+ | 21 | 13 | 19 | 3 | <0.001 (0.603) |
| Very low | 5 | 1 | 0 | 0 | |
| Low | 10 | 5 | 2 | 0 | |
| Intermediate | 4 | 4 | 3 | 0 | |
| High | 2 | 3 | 8 | 1 | |
| Very High | 0 | 0 | 6 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-M.; Lee, H.; Eom, K.-S.; Lee, S.-E.; Kim, M.; Kim, Y. Impact of Integrated Genetic Information on Diagnosis and Prognostication for Myeloproliferative Neoplasms in the Next-Generation Sequencing Era. J. Clin. Med. 2021, 10, 1033. https://doi.org/10.3390/jcm10051033
Lee J-M, Lee H, Eom K-S, Lee S-E, Kim M, Kim Y. Impact of Integrated Genetic Information on Diagnosis and Prognostication for Myeloproliferative Neoplasms in the Next-Generation Sequencing Era. Journal of Clinical Medicine. 2021; 10(5):1033. https://doi.org/10.3390/jcm10051033
Chicago/Turabian StyleLee, Jong-Mi, Howon Lee, Ki-Seong Eom, Sung-Eun Lee, Myungshin Kim, and Yonggoo Kim. 2021. "Impact of Integrated Genetic Information on Diagnosis and Prognostication for Myeloproliferative Neoplasms in the Next-Generation Sequencing Era" Journal of Clinical Medicine 10, no. 5: 1033. https://doi.org/10.3390/jcm10051033