
Gene Expression Landscape of SDH-Deficient Gastrointestinal Stromal Tumors

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
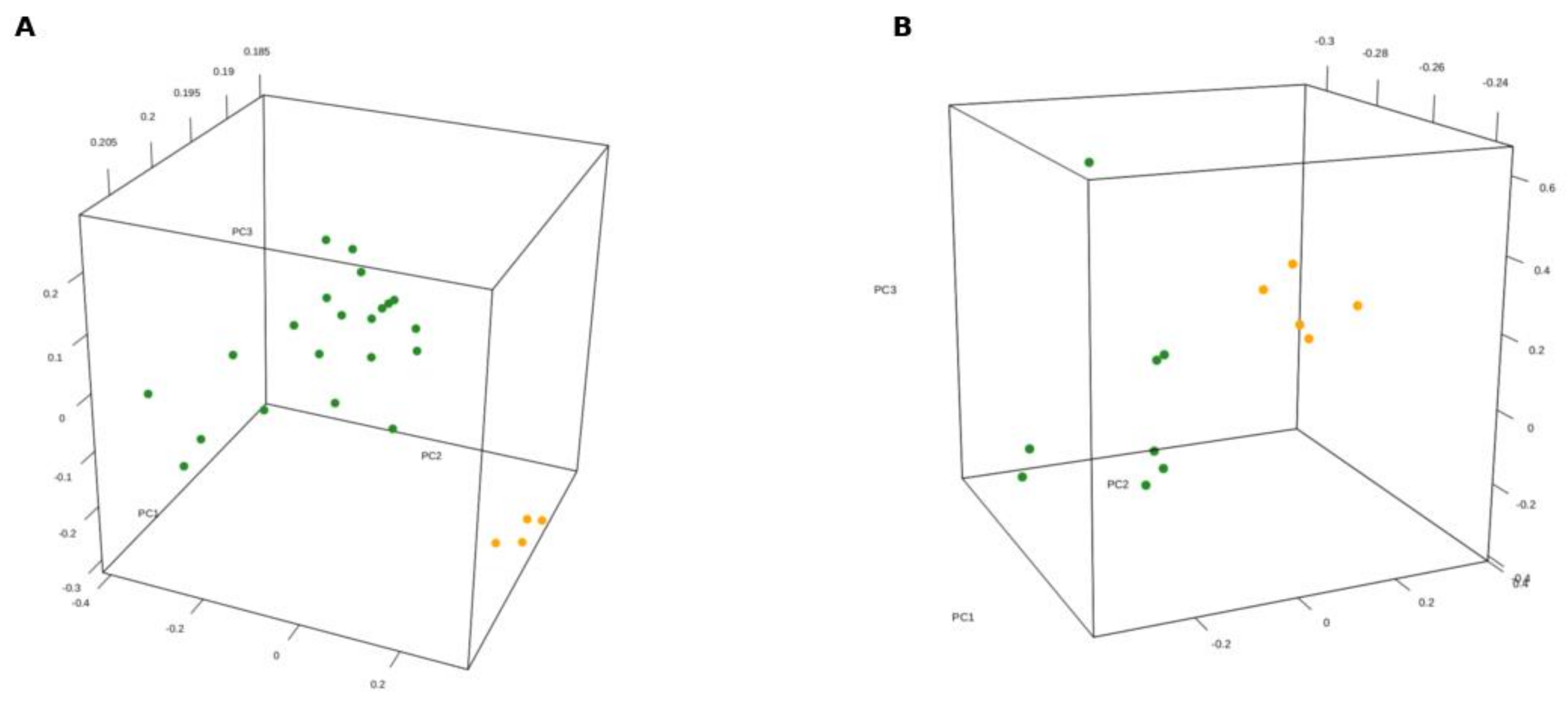
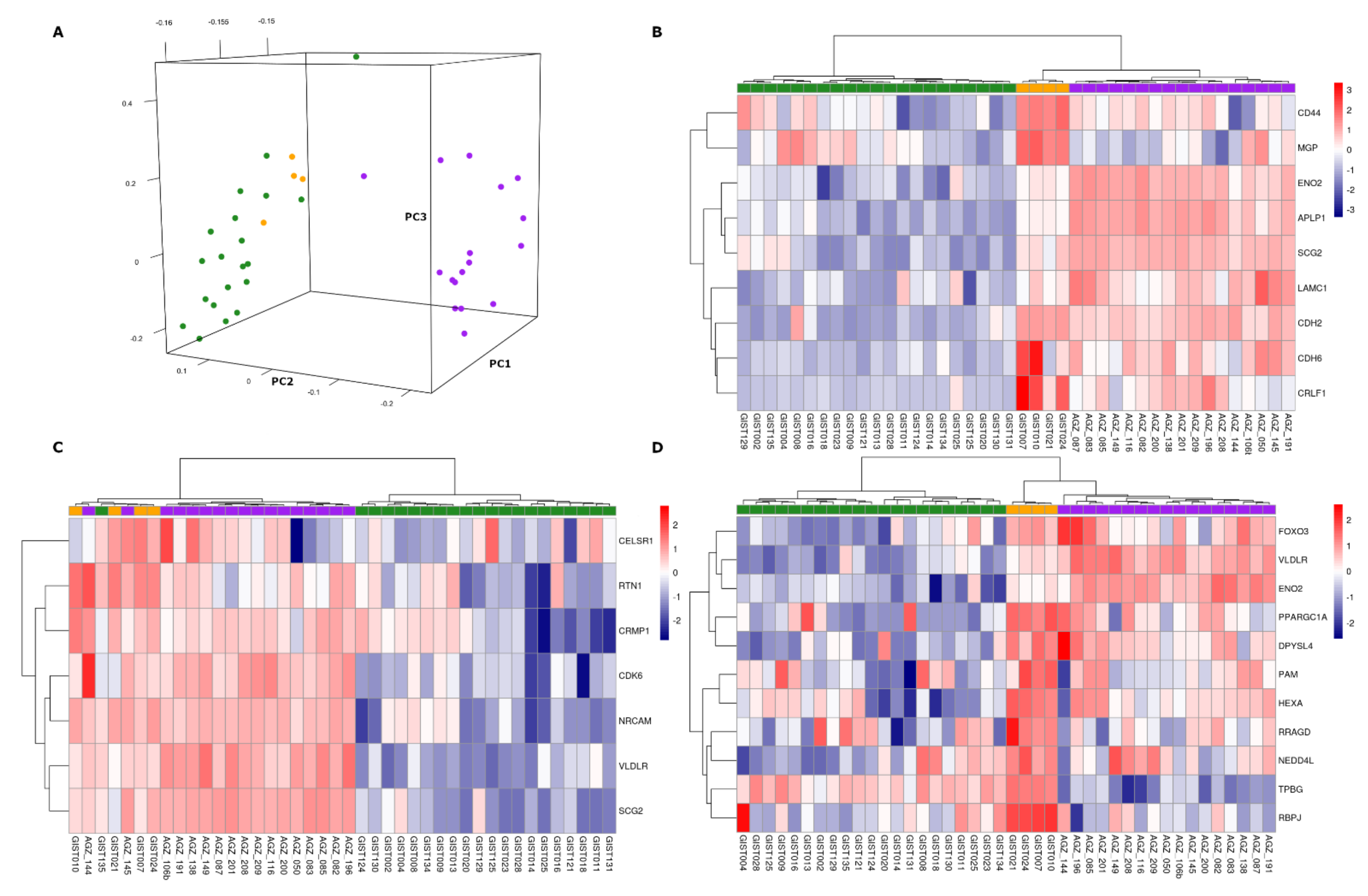
3.1. Gene Expression Profile of SDH-Deficient GIST
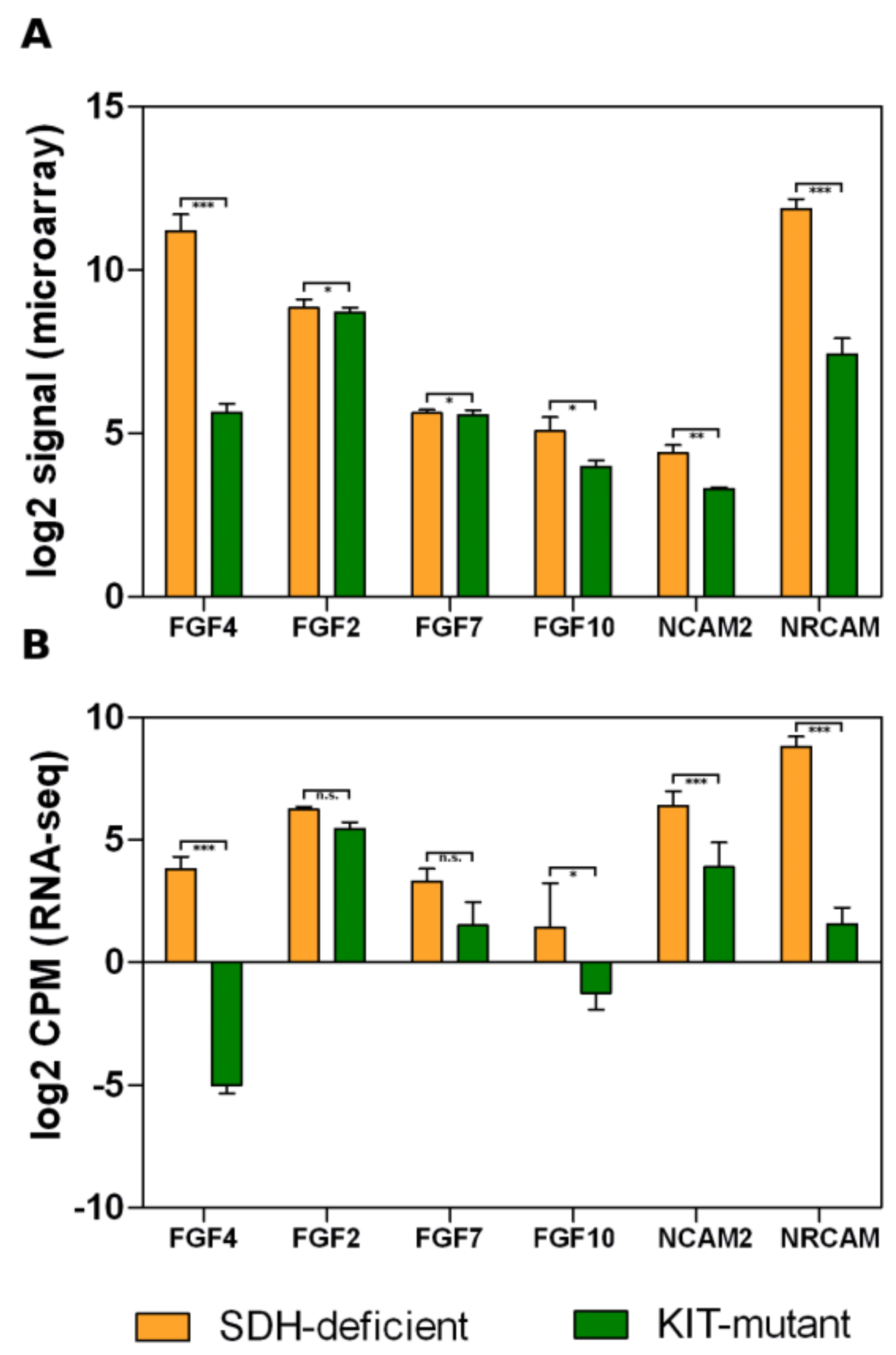
3.2. Overexpression of Neural Markers
3.3. Fibroblast Growth Factor Receptor 2 Binding and Activation
3.4. Comparison with SDH-Deficient Pheochromocytoma and Paraganglioma
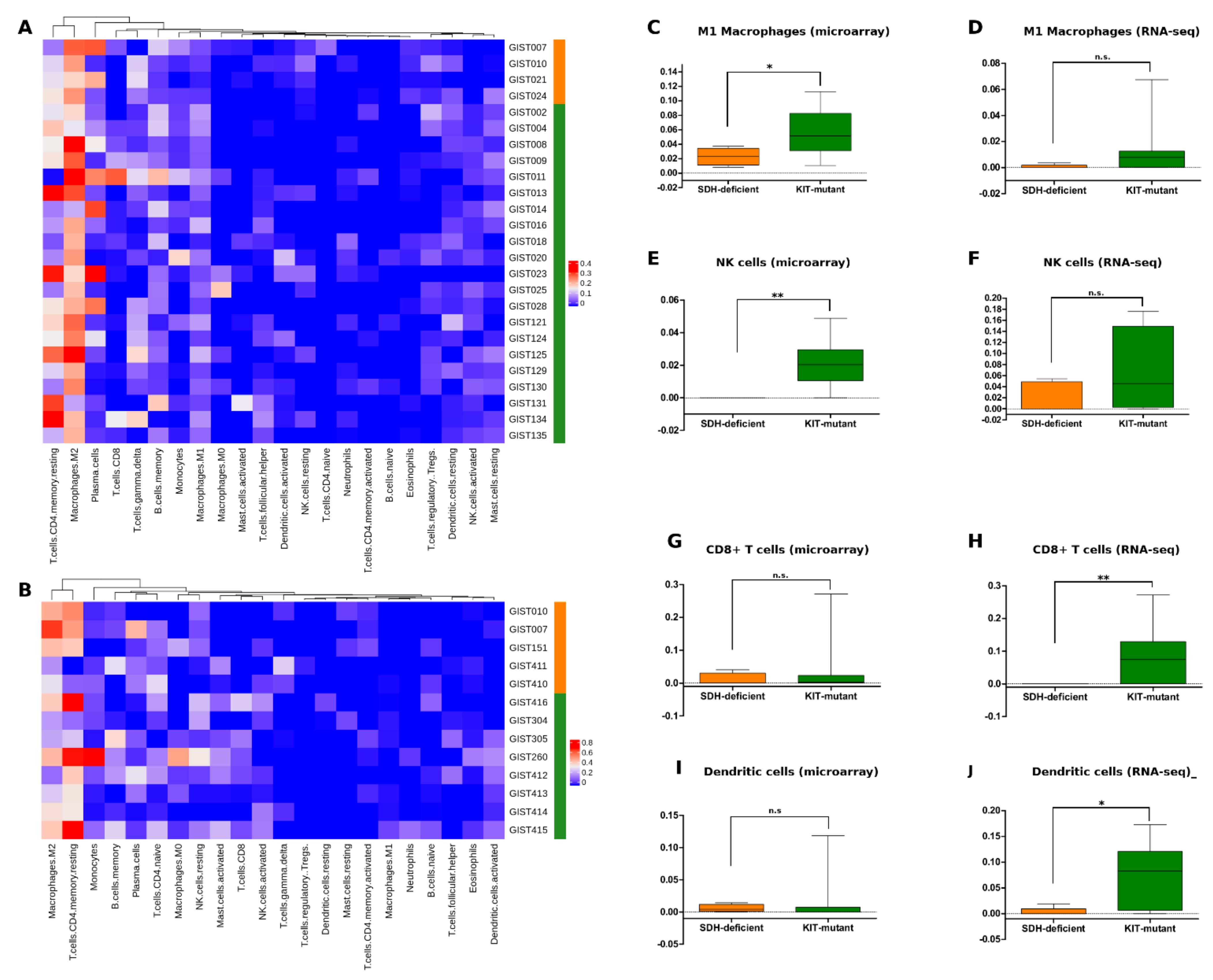
3.5. SDH-Deficient GIST Immune Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in Succinate Dehydrogenase in Gastrointestinal Stromal Tumors Lacking KIT and PDGFRA Mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.-X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of Expression of SDHA Predicts SDHA Mutations in Gastrointestinal Stromal Tumors. Mod. Pathol. 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of All Subunits, SDHA, SDHB, SDHC, SDHD, of the Succinate Dehydrogenase Complex in KIT/PDGFRA Wild-Type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, C.A.; Carney, J.A. The Triad of Paragangliomas, Gastric Stromal Tumours and Pulmonary Chondromas (Carney Triad), and the Dyad of Paragangliomas and Gastric Stromal Sarcomas (Carney-Stratakis Syndrome): Molecular Genetics and Clinical Implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nannini, M.; Urbini, M.; Astolfi, A.; Biasco, G.; Pantaleo, M.A. The Progressive Fragmentation of the KIT/PDGFRA Wild-Type (WT) Gastrointestinal Stromal Tumors (GIST). J. Transl. Med. 2017, 15, 113. [Google Scholar] [CrossRef]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.-M.; Otto, C.; et al. Aberrant DNA Hypermethylation of SDHC: A Novel Mechanism of Tumor Development in Carney Triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Lolli, C.; Nannini, M.; Astolfi, A.; Indio, V.; Saponara, M.; Urbini, M.; La Rovere, S.; Gill, A.; Goldstein, D.; et al. Good Survival Outcome of Metastatic SDH-Deficient Gastrointestinal Stromal Tumors Harboring SDHA Mutations. Genet. Med. 2015, 17, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Chopra, S. Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Arch. Pathol. Lab. Med. 2020, 144, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq Gene Expression Estimation with Read Mapping Uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune Microenvironment Profiling of Gastrointestinal Stromal Tumors (GIST) Shows Gene Expression Patterns Associated to Immune Checkpoint Inhibitors Response. Oncoimmunology 2019, 8, e1617588. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; Nannini, M.; Ceccarelli, C.; Formica, S.; Santini, D.; Heinrich, M.C.; Corless, C.; Dei Tos, A.P.; Paterini, P.; et al. Differential Expression of Neural Markers in KIT and PDGFRA Wild-Type Gastrointestinal Stromal Tumours. Histopathology 2011, 59, 1071–1080. [Google Scholar] [CrossRef]
- Lee, M.Y.; Ha, S.E.; Park, C.; Park, P.J.; Fuchs, R.; Wei, L.; Jorgensen, B.G.; Redelman, D.; Ward, S.M.; Sanders, K.M.; et al. Transcriptome of Interstitial Cells of Cajal Reveals Unique and Selective Gene Signatures. PLoS ONE 2017, 12, e0176031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, N.; Goldstein, A.M. Endothelin-3 Regulates Neural Crest Cell Proliferation and Differentiation in the Hindgut Enteric Nervous System. Dev. Biol. 2006, 293, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.J.; Ma, S.P.; Ju, F.; Zhang, Y.P.; Li, Z.C.; Zhang, B.B.; Lian, J.J.; Wang, L.; Cheng, B.F.; Wang, M.; et al. Thrombospondin-4 Promotes Neuronal Differentiation of NG2 Cells via the ERK/MAPK Pathway. J. Mol. Neurosci. 2016, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, G.; Radenkovic, D.; Velickov, A. Development of Interstitial Cells of Cajal in the Human Digestive Tract as the Result of Reciprocal Induction of Mesenchymal and Neural Crest Cells. J. Cell Mol. Med. 2018, 22, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, A.; Szura, M.; Gil, K.; Matyja, A. Interstitial Cells of Cajal—Systematic Review. Folia Morphol. 2016, 75, 281–286. [Google Scholar] [CrossRef]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The Binding of NCAM to FGFR1 Induces a Specific Cellular Response Mediated by Receptor Trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef]
- Loriot, C.; Burnichon, N.; Gadessaud, N.; Vescovo, L.; Amar, L.; Libé, R.; Bertherat, J.; Plouin, P.-F.; Jeunemaitre, X.; Gimenez-Roqueplo, A.-P.; et al. Epithelial to Mesenchymal Transition Is Activated in Metastatic Pheochromocytomas and Paragangliomas Caused by SDHB Gene Mutations. J. Clin. Endocrinol. Metab. 2012, 97, E954–E962. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Hu, H.; Takano, N.; Xiang, L.; Gilkes, D.M.; Luo, W.; Semenza, G.L. Hypoxia-Inducible Factors Enhance Glutamate Signaling in Cancer Cells. Oncotarget 2014, 5, 8853–8868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiello, G.A.; Bowler, T.G.; Liu, M.; Medina, B.D.; Zhang, J.Q.; Param, N.J.; Loo, J.K.; Goldfeder, R.L.; Chibon, F.; Rossi, F.; et al. Differential Immune Profiles Distinguish the Mutational Subtypes of Gastrointestinal Stromal Tumor. J. Clin. Investig. 2019, 129, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-Related MRNA Profile Predicts Clinical Response to PD-1 Blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-Cancer Adaptive Immune Resistance as Defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Indio, V.; Ravegnini, G.; Astolfi, A.; Urbini, M.; Saponara, M.; De Leo, A.; Gruppioni, E.; Tarantino, G.; Angelini, S.; Pession, A.; et al. Gene Expression Profiling of PDGFRA Mutant GIST Reveals Immune Signatures as a Specific Fingerprint of D842V Exon 18 Mutation. Front. Immunol. 2020, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Nannini, M.; Astolfi, A.; Urbini, M.; Indio, V.; Santini, D.; Heinrich, M.C.; Corless, C.L.; Ceccarelli, C.; Saponara, M.; Mandrioli, A.; et al. Integrated Genomic Study of Quadruple-WT GIST (KIT/PDGFRA/SDH/RAS Pathway Wild-Type GIST). BMC Cancer 2014, 14, 685. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-Mediated Carcinogenesis. Biochim. Biophys. Acta 2012, 1823, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr Gene Families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int. J. Mol. Sci. 2020, 21, 3313. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered Chromosomal Topology Drives Oncogenic Programs in SDH-Deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Urbini, M.; Astolfi, A.; Indio, V.; Nannini, M.; Schipani, A.; Bacalini, M.G.; Angelini, S.; Ravegnini, G.; Calice, G.; Del Gaudio, M.; et al. Gene Duplication, Rather than Epigenetic Changes, Drives FGF4 Overexpression in KIT/PDGFRA/SDH/RAS-P WT GIST. Sci. Rep. 2020, 10, 19829. [Google Scholar] [CrossRef]
- Ernst, S.; Langer, R.; Cooney, C.L.; Sasisekharan, R. Enzymatic Degradation of Glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387–444. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Morla, S. Glycosaminoglycans and Glycosaminoglycan Mimetics in Cancer and Inflammation. Int. J. Mol. Sci. 2019, 20, 1963. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Røsland, G.V.; Dyrstad, S.E.; Tusubira, D.; Helwa, R.; Tan, T.Z.; Lotsberg, M.L.; Pettersen, I.K.N.; Berg, A.; Kindt, C.; Hoel, F.; et al. Epithelial to Mesenchymal Transition (EMT) Is Associated with Attenuation of Succinate Dehydrogenase (SDH) in Breast Cancer through Reduced Expression of SDHC. Cancer Metab. 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grünewald, T.G.P. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluckova, K.; Tennant, D.A. Metabolic Implications of Hypoxia and Pseudohypoxia in Pheochromocytoma and Paraganglioma. Cell Tissue Res. 2018, 372, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/Pseudohypoxia-Mediated Activation of Hypoxia-Inducible Factor-1α in Cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Günther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T.; et al. Hypoxia and Oxygenation Induce a Metabolic Switch between Pentose Phosphate Pathway and Glycolysis in Glioma Stem-like Cells. Acta Neuropathol. 2013, 126, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Indio, V.; Astolfi, A.; Urbini, M.; Nannini, M.; Pantaleo, M.A. Genetics and Treatment of Gastrointestinal Stromal Tumors with Immune Checkpoint Inhibitors: What Do We Know? Pharmacogenomics 2020, 21, 231–234. [Google Scholar] [CrossRef] [PubMed]
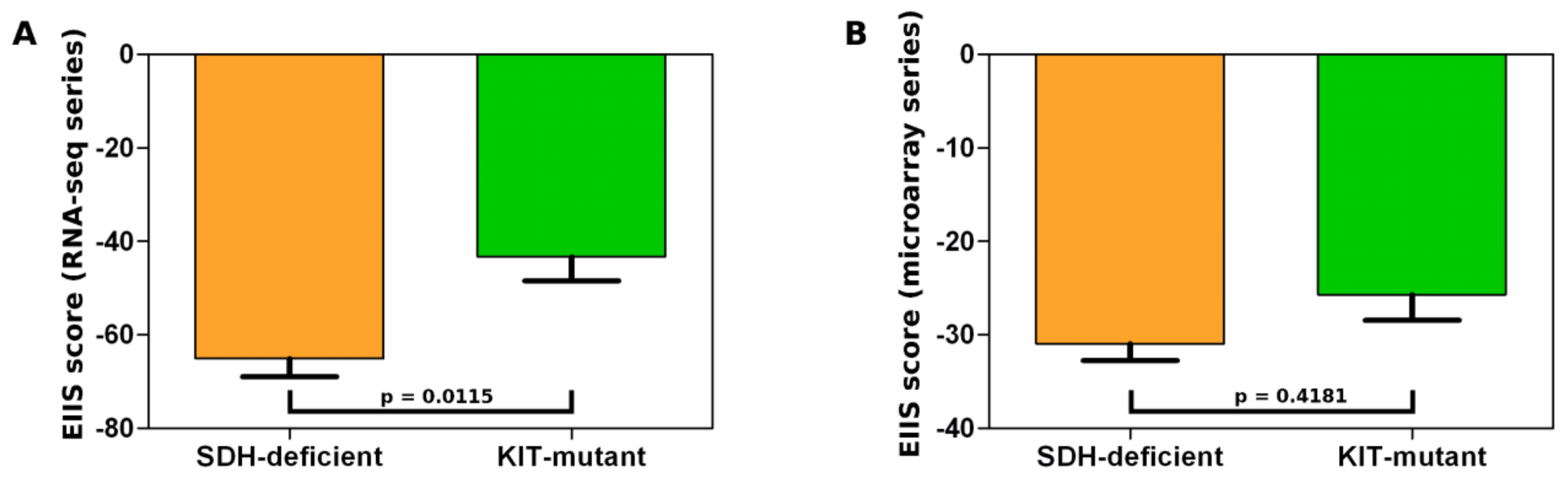


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SDH-Deficient | KIT-Mutant | |
|---|---|---|
| Sex | ||
| Female | 4 | 11 |
| Male | 3 | 18 |
| Age (average, range) | 25 (18–30) | 62.4 (34–87) |
| Site | ||
| Intestine | 0 | 9 |
| Stomach | 6 | 17 |
| NA | 1 | 3 |
| Tumor size | ||
| <5 cm | 0 | 4 |
| ≥5 cm | 5 | 22 |
| NA | 2 | 3 |
| Mitotic rate | ||
| <5/50 HPF * | 0 | 9 |
| ≥5/50 HPF | 4 | 15 |
| NA | 3 | 5 |
| Desease status at diagnosis | ||
| Localized | 0 | 18 |
| Metastatic | 5 | 7 |
| NA | 2 | 4 |
| Platform | ||
| Microarray | 4 ** | 21 |
| RNA-seq | 5 ** | 8 |
| Database | Terms | p-Value |
|---|---|---|
| MSigDB Hallmark up-regulated genes | Hedgehog Signaling | 0.00001 |
| Hypoxia | 0.00263 | |
| UV Response Dn | 0.0027 | |
| Estrogen Response Early | 0.00779 | |
| Glycolysis | 0.00779 | |
| Apical Junction | 0.02104 | |
| Epithelial Mesenchymal Transition | 0.02104 | |
| MSigDB Hallmark down-regulated genes | TNF-alpha Signaling via NF-kB | 0.00001 |
| Interferon Gamma Response | 0.00013 | |
| KRAS Signaling Up | 0.00013 | |
| Fatty Acid Metabolism | 0.00031 | |
| IL-2/STAT5 Signaling | 0.0005 | |
| Complement | 0.0019 | |
| IL-6/JAK/STAT3 Signaling | 0.00322 | |
| Bile Acid Metabolism | 0.01086 | |
| Adipogenesis | 0.01857 | |
| Interferon Alpha Response | 0.02256 | |
| Human Gene Atlas up-regulated genes | Fetalbrain | 0 |
| pineal night | 0.00001 | |
| PrefrontalCortex | 0.00043 | |
| Amygdala | 0.00072 | |
| pineal day | 0.00412 | |
| SuperiorCervicalGanglion | 0.01149 | |
| Thyroid | 0.01317 | |
| CerebellumPeduncles | 0.02167 | |
| BronchialEpithelialCells | 0.02805 | |
| Human Gene Atlas down-regulated genes | CD33+ Myeloid | 0.00018 |
| CD14+ Monocytes | 0.00069 | |
| CD56+ NKCells | 0.00173 | |
| Prostate | 0.00303 | |
| Liver | 0.0151 | |
| BronchialEpithelialCells | 0.01868 | |
| Adipocyte | 0.03136 |
| Microarray | RNA-seq | |||
|---|---|---|---|---|
| Pathway NAME | NES * | p-Value | NES | p-Value |
| REACTOME PHOSPHOLIPASE C MEDIATED CASCADE FGFR2 | 1.802 | 0.022222 | 1.354 | 0 |
| REACTOME_FGFR2_LIGAND_BINDING_AND_ACTIVATIONC ** | 1.717 | 0.044444 | 1.313 | 0.0625 |
| WP GLYCOSAMINOGLYCAN DEGRADATION | 1.686 | 0.041667 | 1.346 | 0.02564 |
| REACTOME VXPX CARGO TARGETING TO CILIUM | 1.576 | 0.019231 | 1.276 | 0.040816 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indio, V.; Schipani, A.; Nannini, M.; Urbini, M.; Rizzo, A.; De Leo, A.; Altimari, A.; Di Scioscio, V.; Messelodi, D.; Tarantino, G.; et al. Gene Expression Landscape of SDH-Deficient Gastrointestinal Stromal Tumors. J. Clin. Med. 2021, 10, 1057. https://doi.org/10.3390/jcm10051057
Indio V, Schipani A, Nannini M, Urbini M, Rizzo A, De Leo A, Altimari A, Di Scioscio V, Messelodi D, Tarantino G, et al. Gene Expression Landscape of SDH-Deficient Gastrointestinal Stromal Tumors. Journal of Clinical Medicine. 2021; 10(5):1057. https://doi.org/10.3390/jcm10051057
Chicago/Turabian StyleIndio, Valentina, Angela Schipani, Margherita Nannini, Milena Urbini, Alessandro Rizzo, Antonio De Leo, Annalisa Altimari, Valerio Di Scioscio, Daria Messelodi, Giuseppe Tarantino, and et al. 2021. "Gene Expression Landscape of SDH-Deficient Gastrointestinal Stromal Tumors" Journal of Clinical Medicine 10, no. 5: 1057. https://doi.org/10.3390/jcm10051057
APA StyleIndio, V., Schipani, A., Nannini, M., Urbini, M., Rizzo, A., De Leo, A., Altimari, A., Di Scioscio, V., Messelodi, D., Tarantino, G., Astolfi, A., & Pantaleo, M. A. (2021). Gene Expression Landscape of SDH-Deficient Gastrointestinal Stromal Tumors. Journal of Clinical Medicine, 10(5), 1057. https://doi.org/10.3390/jcm10051057