
Partial Lipodystrophy and LMNA p.R545H Variant

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
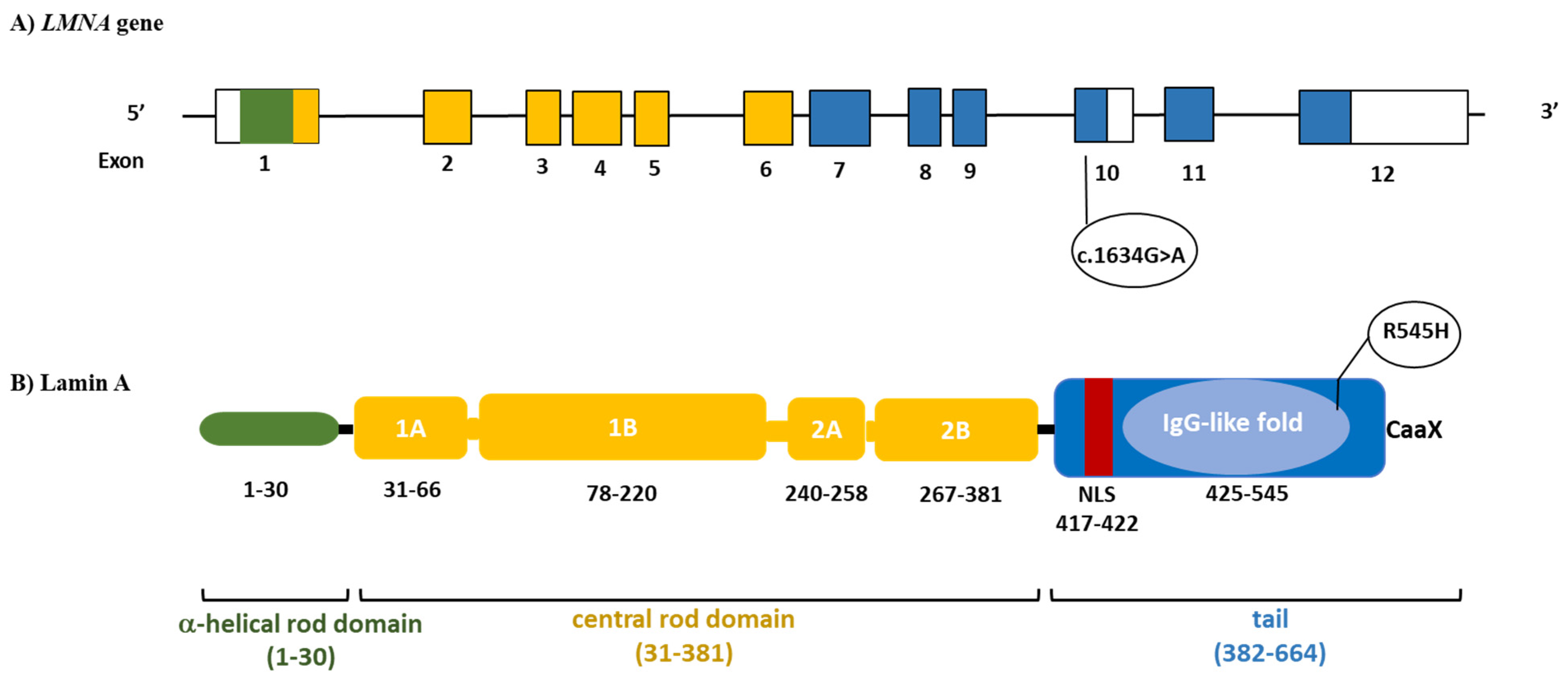
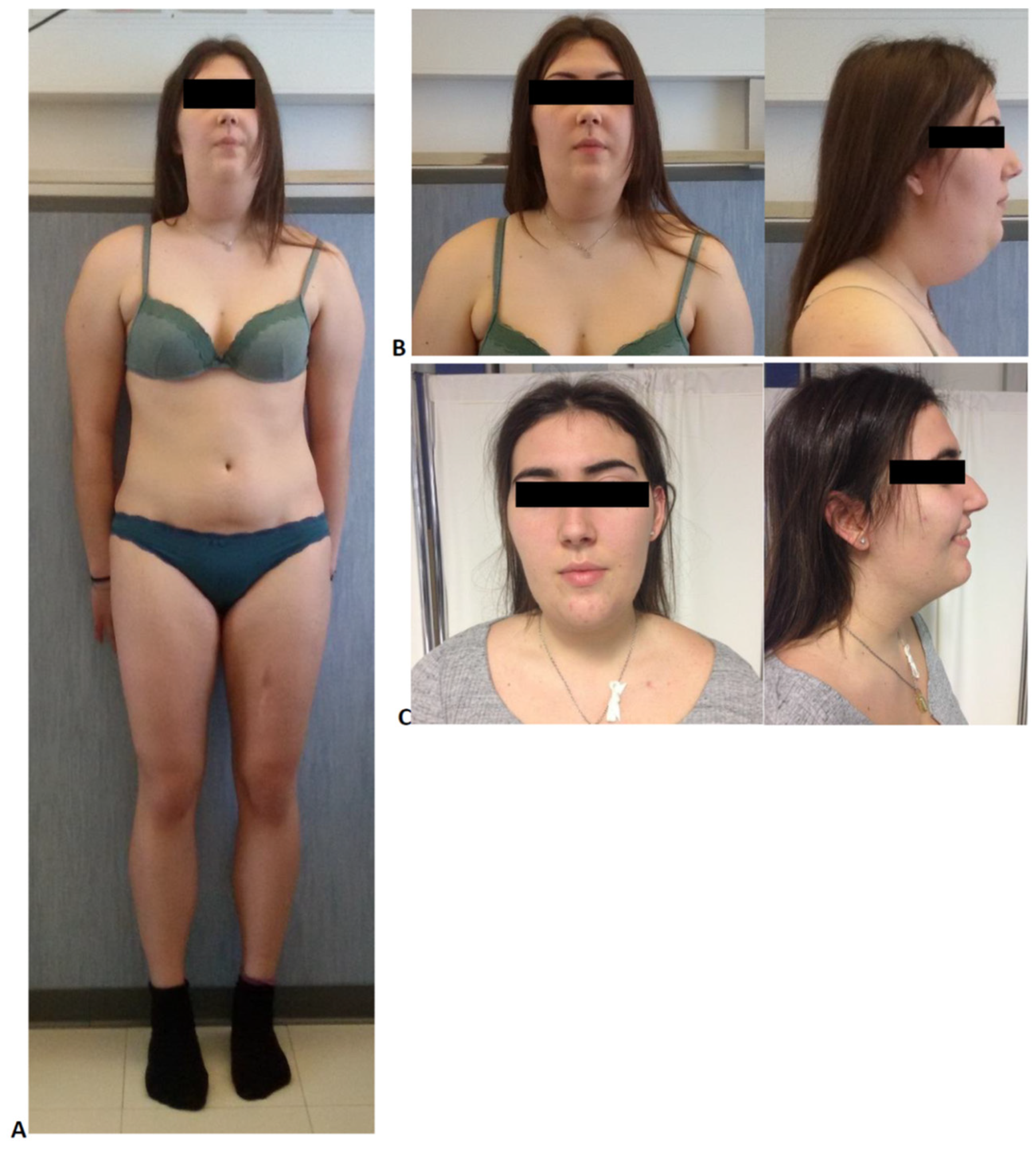
2. Case Description
3. Methods
3.1. Anthropometric Measurements
3.2. Biochemistry and Hormones
3.3. Genetic Testing
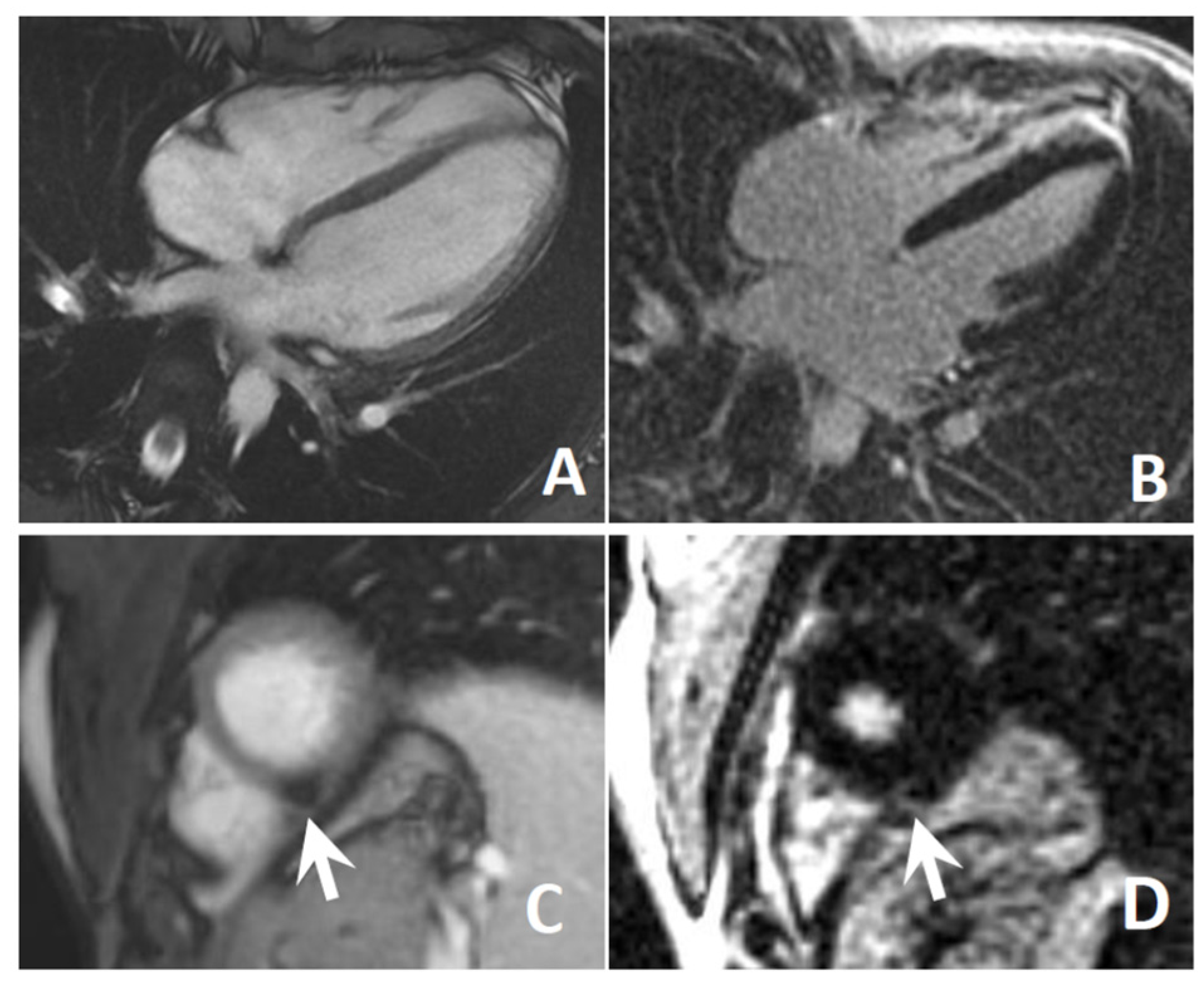
3.4. Cardiological Assessment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Araújo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, G.; Benedetti, S.; D’Apice, M.R.; Maggi, L.; Carboni, N.; Scarano, E.; Politano, L. Emergingperspectives on laminopathies. Cell Health Cytoskelet. 2016, 8, 25–35. [Google Scholar] [CrossRef]
- Magno, S.; Ceccarini, G.; Pelosini, C.; Ferrari, F.; Prodam, F.; Gilio, D.; Maffei, M.; Sessa, M.R.; Barison, A.; Ciccarone, A. Atypical Progeroid Syndrome and Partial Lipodystrophy Due to LMNA Gene p.R349W Mutation. J. Endocr. Soc. 2020, 4, bvaa108. [Google Scholar] [CrossRef]
- Carboni, N.; Politano, L.; Floris, M.; Mateddu, A.; Solla, E.; Olla, S.; Maggi, L.; Maioli, M.A.; Piras, R.; Cocco, E.; et al. Overlapping syndromes in laminopathies: A meta-analysis of the reported literature. Acta Myol. 2013, 32, 7–17. [Google Scholar]
- Dittmer, T.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.; Eriksson, M. Low and High Expressing Alleles of the LMNA Gene: Implications for Laminopathy Disease Development. PLoS ONE 2011, 6, e25472. [Google Scholar] [CrossRef]
- Hegele, R. LMNA mutation position predicts organ system involvement in laminopathies. Clin. Genet. 2005, 68, 31–34. [Google Scholar] [CrossRef]
- Lin, E.W.; Brady, G.F.; Kwan, R.; Nesvizhskii, A.I.; Omary, M.B. Genotype-phenotype analysis of LMNA-related diseases predicts phenotype-selective alterations in lamin phosphorylation. FASEB J. 2020, 1–23. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Sánchez-Iglesias, S.; Mera, A.; Pintos, E.; Castro-Pais, A.; Rodríguez-Cañete, L.; Pardo, J.; Casanueva, F.F.; Araújo-Vilar, D. Inflammatory myopathy in the context of an unusual overlapping laminopathy. Arch. Endocrinol. Metab. 2018, 62, 376–382. [Google Scholar] [CrossRef]
- Chan, D.; McIntyre, A.D.; Hegele, R.A.; Don-Wauchope, A.C. Familial partial lipodystrophy presenting as metabolic syndrome. J. Clin. Lipidol. 2016, 10, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Patni, N.; Hatab, S.; Xing, C.; Zhou, Z.; Quittner, C.; Garg, A. A novel autosomal recessive lipodystrophy syndrome due to homozygous LMNA variant. J. Med. Genet. 2020, 57, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Vasandani, C.; Li, X.; Sekizkardes, H.; Adams-Huet, B.; Brown, R.J.; Garg, A. Diagnostic Value of Anthropometric Measurements for Familial Partial Lipodystrophy, Dunnigan Variety. J. Clin. Endocrinol. Metab. 2020, 105, 2132–2141. [Google Scholar] [CrossRef]
- Garg, A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 2000, 85, 1776–1782. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 233–270. [Google Scholar] [CrossRef]
- Barison, A.; Aimo, A.; Mirizzi, G.; Castiglione, V.; Ripoli, A.; Panchetti, L.; Rossi, A.; Giannoni, A.; Startari, U.; Aquaro, G.D.; et al. The extent and location of late gadolinium enhancement predict defibrillator shock and cardiac mortality in patients with non-ischaemic dilated cardiomyopathy. Int. J. Cardiol. 2020, 15, 180–186. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Fernández-Pombo, A.; Sánchez-Iglesias, S.; Araújo-Vilar, D. Lipodystrophic laminopathies: Diagnostic clues. Nucleus 2018, 9, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef] [PubMed]
- Kandert, S.; Wehnert, M.; Müller, C.R.; Buendia, B.; Dabauvalle, M.C. Impaired nuclear functions lead to increased senescence and inefficient differentiation in human myoblasts with a dominant p.R545C mutation in the LMNA gene. Eur. J. Cell Biol. 2009, 88, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Vytopil, M.; Benedetti, S.; Ricci, E.; Galluzzi, G.; Dello Russo, A.; Merlini, L.; Boriani, G.; Gallina, M.; Morandi, L.; Politano, L.; et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J. Med. Genet. 2003, 40, e132. [Google Scholar] [CrossRef] [PubMed]
- Decaudain, A.; Vantyghem, M.C.; Guerci, B.; Hécart, A.C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.H.; Donadille, B.; Lebbé, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef]
- Subramanyam, L.; Simha, V.; Garg, A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin. Genet. 2010, 78, 66–73. [Google Scholar] [CrossRef]
- Mory, P.B.; Crispim, F.; Freire, M.B.; Salles, J.E.; Valério, C.M.; Godoy-Matos, A.F.; Dib, S.A.; Moisés, R.S. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. Eur. J. Endocrinol. 2012, 167, 423–431. [Google Scholar] [CrossRef]
- Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Huong, S. Molecular Genetic Studies in Hereditary Laminopathies of Man. Ph.D. Thesis, Ernst-Moritz-Arndt-Universität, Greifswald, Germany, 2010. [Google Scholar]
- van Rijsingen, I.A.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, P.; van den Berg, M.P.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail. 2013, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Olaopa, M.A.; Spoonamore, K.G.; Bhakta, D.; Chen, Z.; Celestino-Soper, P.B.; Chen, P.S.; Ai, T.; Vatta, M. Lamin-A/C variants found in patients with cardiac conduction disease reduce sodium currents. Cardiogenetics 2018, 8. [Google Scholar] [CrossRef]
- van Berlo, J.H.; De Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Le Dour, C.; Schneebeli, S.; Bakiri, F.; Darcel, F.; Jacquemont, M.L.; Maubert, M.A.; Auclair, M.; Jeziorowska, D.; Reznik, Y.; Béréziat, V.; et al. A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: New insights into the pathogenicity of nonfarnesylated prelamin-A. J. Clin. Endocrinol. Metab. 2011, 96, E856–E862. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, R.M.; Costa-Riquetto, A.D.; Fernandes, V.O.; Montenegro, A.; de Santana, L.S.; Jorge, A.A.L.; Karbage, L.; Aguiar, L.B.; Carvalho, F.H.C.; Teles, M.G.; et al. Homozygous and Heterozygous Nuclear Lamin A p.R582C Mutation: Different Lipodystrophic Phenotypes in the Same Kindred. Front. Endocrinol. 2018, 9, 458. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Magré, J.; Vantyghem, M.C.; Bourut, C.; Lascols, O.; Shackleton, S.; Lloyd, D.J.; Guerci, B.; Padova, G.; Valensi, P.; et al. Lamin A/C gene: Sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000, 49, 1958–1962. [Google Scholar] [CrossRef]
- Von Schnurbein, J.; Adams, C.; Akinci, B.; Ceccarini, G.; D’Apice, M.R.; Gambineri, A.; Hennekam, R.C.M.; Jeru, I.; Lattanzi, G.; Miehle, K.; et al. European lipodystrophy registry: Background and structure. Orphanet. J. Rare Dis. 2020, 15, 17. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Index Case | Father of the Index Case | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| Current Study | Current Study | [10] | [11] | [12] | [12] | [12] | [12] | |
| Sex | F | M | F | F | F | M | F | F |
| LMNA pR545H | HT | HT | HT | HT | HT | HT | HO | HO |
| Age at report (y) | 21 | 45 | 48 | 51 | 40 | 16 | 16 | 16 |
| Height (m) | 1.72 | 1.74 | 1.61 | 1.62 | 1.55 | 1.64 | 1.40 | 1.44 |
| Weight (kg) | 71 | 72 | 65.3 | 61.1 | 96.8 | 100.2 | 32.2 | 37.8 |
| BMI (kg/m2) | 24 | 23.8 | 25.2 | 23.1 | 40.2 | 36.9 | 16.4 | 18.1 |
| Lipoatrophy | - | - | Mild (limbs and buttocks) | Mild (limbs and buttocks) | - | - | Severe (Trunk limbs and buttocks) | Severe (Trunk limbs and buttocks) |
| Fat Accumulation | Neck | - | Face/Neck/Abdomen | Neck/Abdomen | Obese | Obese | - | - |
| Acanthosis Nigricans | - | - | NR | NR | + | + | - | NR |
| Hepatic steatosis | - | - | NR | + | NR | NR | + | + |
| Hypertriglyceridemia | - | - | + | + | - | - | + | + |
| Diabetes/IR | - | + | + | + | - | + | + | |
| Cardiovascular Involvement | - | - | WPW syndrome | Hypertension | - | - | NR | NR |
| Myopathy | + | - | + | - | - | - | - | NR |
| Additional characteristics | - | - | - | - | - | Umblical hernia, orchidopexy for UDT | Intellectual disability, clinodactyly, joint contractures, cataracts, uterine leiomyoma | Intellectual disability, clinodactyly, joint contractures, cataracts, uterine leiomyoma |
| Whole body fat (%) | 31.5 | 20.9 | 30.6 | - | 39.9 | - | 22.3 | 26.8 |
| Arm fat (%) | 43 | 17.9 | 31.6 | - | 55.9 | - | 25 | 27.6 |
| Leg fat (%) | 33.9 | 20.8 | 30.5 | - | 34.1 | - | 17 | 16.5 |
| Truncal fat (%) | 26.8 | 22.3 | 31.8 | - | 39.2 | - | 24.1 | 32.2 |
| Skinfold thickness | ||||||||
| Abdomen (mm) | 21 | 22 | - | - | - | - | 12 | 16 |
| Suprailiac (mm) | 9 | 15 | 15 | - | - | - | 10 | 12 |
| Subscapular (mm) | 15 | 16 | 18 | - | - | - | 7 | 11 |
| Biceps (mm) | 11 | 5 | 10 | - | - | - | 2 | 6 |
| Triceps (mm) | 18 | 12 | 17 | - | - | - | 4 | 4 |
| Midthigh (mm) | 16 | 13 | 10 | - | - | - | 6 | 5 |
| Calf (mm) | 10 | 7 | 5 | - | - | - | 3 | 6 |
| Index Case | Father of the Index Case | * Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| Current Study | Current Study | [10] | [11] | [12] | [12] | [12] | [12] | |
| Glucose | ||||||||
| Fasting (mg/dL) | 85 | 84 | 65–91 | 99 | 85 | 83 | 292 | 146 |
| 1 h OGTT (mg/dL) | 136 | 139 | - | NR | - | - | - | - |
| 2 h OGTT (mg/dL) | 94 | 102 | - | 229 | - | - | - | - |
| HbA1c (%) | 5 | 5.4 | 6–6.1 | NR | 6.1 | 5 | 7.2 | 6.7 |
| Insulin | ||||||||
| Fasting (µUI/mL) | 9.76 | 2.62 | 43.9 | NR | NR | NR | NR | NR |
| 1 h OGTT (µUI/mL) | 70.1 | 40.7 | - | NR | - | - | - | - |
| 2 h OGTT(µUI/mL) | 38.8 | 20.5 | - | NR | - | - | - | - |
| Lipids | ||||||||
| Total-C (mg/dL) | 161 | 176 | 63–139 | 319 | 208 | 165 | >1000 | 210 |
| HDL-C (mg/dL) | 54 | 73 | 40–40 | 42 | 51 | 37 | 10 | 37 |
| LDL-C (mg/dL) | 100 | 113 | NR | NR | 185 | 109 | - | - |
| TG (mg/dL) | 65 | 54 | 202–214 | 230 | 185 | 99 | 5436 | 262 |
| Liver function | ||||||||
| AST (U/L) | 38 | 15 | NR | NR | 18 | 20 | 60 | 45 |
| ALT (U/L) | 25 | 17 | NR | 39 | 17 | 21 | 111 | 77 |
| γGT(U/L) | 10 | 13 | NR | NR | NR | NR | NR | NR |
| CPK (U/L) | 390-46-70 | 88 | 378–2500 | 50 | NR | NR | NR | NR |
| LDH (U/L) | 221 | 157 | NR | NR | NR | NR | NR | NR |
| Leptin (ng/mL) | 10.4 | 1.4 | 1–8.4 | NR | NR | NR | 1.9 | 9.1 |
| Adiponectin (mcg/mL) | 5 | - | NR | NR | NR | NR | <2 | 2 |
| In Silico Tool | Score | Prediction | Basis Algorithm |
|---|---|---|---|
| Polyphen-2 1 | 0.999 | Probably Damaging | Protein structure/function and evolutionary conservation |
| Mutation Taster 2 | 29 | Disease Causing | Protein structure/function and evolutionary conservation |
| CADD 3 | 26.2 | Very Likely Deleterious | Contrasts annotations of fixed/nearly fixed derived alleles in humans with simulated variants |
| FATHMM 4 | −4.90 | Damaging | Evolutionary conservation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magno, S.; Ceccarini, G.; Barison, A.; Fabiani, I.; Giacomina, A.; Gilio, D.; Pelosini, C.; Rubegni, A.; Emdin, M.; Gatti, G.L.; et al. Partial Lipodystrophy and LMNA p.R545H Variant. J. Clin. Med. 2021, 10, 1142. https://doi.org/10.3390/jcm10051142
Magno S, Ceccarini G, Barison A, Fabiani I, Giacomina A, Gilio D, Pelosini C, Rubegni A, Emdin M, Gatti GL, et al. Partial Lipodystrophy and LMNA p.R545H Variant. Journal of Clinical Medicine. 2021; 10(5):1142. https://doi.org/10.3390/jcm10051142
Chicago/Turabian StyleMagno, Silvia, Giovanni Ceccarini, Andrea Barison, Iacopo Fabiani, Alessandro Giacomina, Donatella Gilio, Caterina Pelosini, Anna Rubegni, Michele Emdin, Gian Luca Gatti, and et al. 2021. "Partial Lipodystrophy and LMNA p.R545H Variant" Journal of Clinical Medicine 10, no. 5: 1142. https://doi.org/10.3390/jcm10051142
APA StyleMagno, S., Ceccarini, G., Barison, A., Fabiani, I., Giacomina, A., Gilio, D., Pelosini, C., Rubegni, A., Emdin, M., Gatti, G. L., Santorelli, F. M., Sessa, M. R., & Santini, F. (2021). Partial Lipodystrophy and LMNA p.R545H Variant. Journal of Clinical Medicine, 10(5), 1142. https://doi.org/10.3390/jcm10051142