
Thrombocytopathies: Not Just Aggregation Defects—The Clinical Relevance of Procoagulant Platelets

 ,
,  ,
, 
Abstract
:1. Introduction
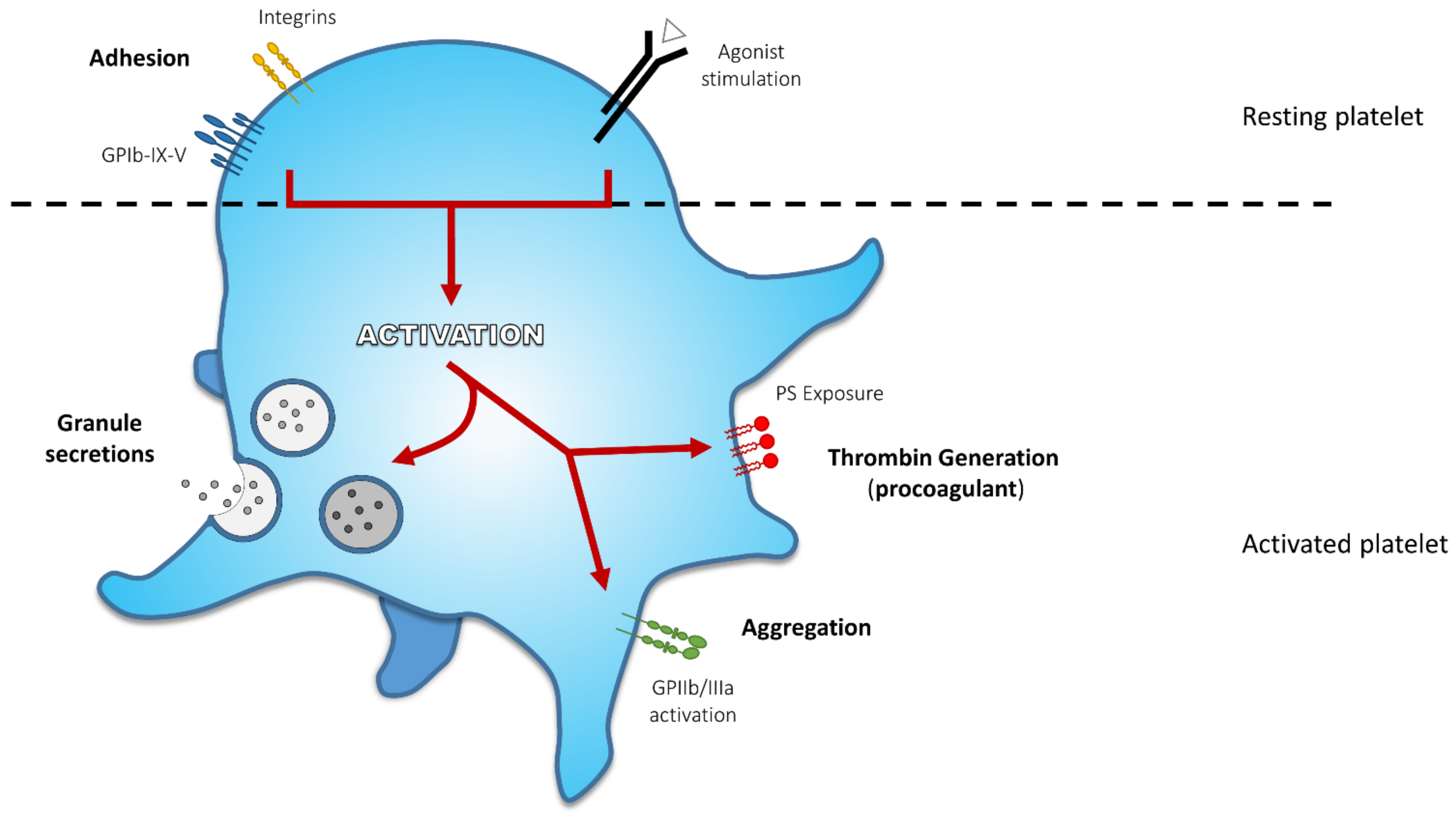
2. Platelet Activation End-Points and Related Defects
2.1. Adhesion
2.1.1. Bernard-Soulier Syndrome
2.1.2. Platelet Type von Willebrand’s Disease
2.2. Secretion
2.2.1. α-Storage Pool Disease or Gray Platelet Syndrome
2.2.2. δ-Storage Pool Disease
2.3. Aggregation
Glanzmann Thrombasthenia
- -
- ߓType I, the most severe form of GT: the expression of GPIIb/IIIa is absent (<5% of normal); platelet fibrinogen and clot retraction are also absent;
- -
- ߓType II, a moderate form of the disease: surface GPIIb/IIIa is reduced with a level of expression varying between 10–20% of normal; reduced fibrinogen content and clot retraction;
- -
- ߓType III, a variant form: the expression of GPIIb/IIIa is near normal or normal (between 50–100%), but the receptor is dysfunctional; variable platelet fibrinogen content and clot retraction.
2.4. Procoagulant Activity
3. Expression of Negatively Charged Phospholipids and Their Role in Coagulation
4. Procoagulant Platelets
4.1. Clinical Features of Procoagulant Platelets
4.1.1. Low Level of Procoagulant Platelets Is Associated with Impaired Platelet Function and Bleeding Diathesis
4.1.2. High Level of Procoagulant Platelets Worsens Thrombotic Events
4.1.3. Procoagulant Platelets in Non-Haemostatic Pathologies
4.2. Pharmacological Modulation of Procoagulant Platelets
4.2.1. Antiplatelet Drugs
4.2.2. Off-Target Procoagulant Platelet Modulation
4.3. Laboratory Work-Up for Investigating Procoagulant Platelets
4.3.1. Quantification and Characterization of Procoagulant Platelets
4.3.2. Assessment of the Overall Coagulation Potential and Procoagulant Activity of Platelets
4.3.3. In-Vivo Investigations of Procoagulant Platelets
5. Thrombocytopathy Associated to COVID-19
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Jobe, S.M.; Di Paola, J. Congenital and acquired disorders of platelet function and number. In Consultative Hemostasis and Thrombosis; Elsevier: Amsterdam, The Netherlands, 2019; pp. 145–166. [Google Scholar]
- Shen, Y.M.; Frenkel, E.P. Acquired platelet dysfunction. Hematol. Oncol. Clin. North. Am. 2007, 21, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Cherry-Bukowiec, J.; Napolitano, L. What platelet disorders occur in the intensive care unit and how should they be treated? In Evidence-Based Practice of Critical Care; Elsevier: Amsterdam, The Netherlands, 2010; pp. 645–660. [Google Scholar]
- Bye, A.P.; Unsworth, A.J.; Gibbins, J.M. Platelet signaling: A complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. 2016, 14, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Stegner, D.; Nieswandt, B. Platelet receptor signaling in thrombus formation. J. Mol. Med. 2011, 89, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, J.W.; Bevers, E.M.; Lindhout, T. Platelet activation and blood coagulation. Thromb. Haemost. 2002, 88, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Andrews, R.K.; Lopez, J.A.; Berndt, M.C. Molecular mechanisms of platelet adhesion and activation. Int. J. Biochem. Cell Biol. 1997, 29, 91–105. [Google Scholar] [CrossRef]
- Lopez, J.A.; Dong, J.F. Structure and function of the glycoprotein ib-ix-v complex. Curr. Opin. Hematol. 1997, 4, 323–329. [Google Scholar] [CrossRef]
- Li, R.; Emsley, J. The organizing principle of the platelet glycoprotein ib-ix-v complex. J. Thromb. Haemost. 2013, 11, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.K.; Berndt, M.C. The gpib-ix-v complex. In Platelets; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 195–213. [Google Scholar]
- Romo, G.M.; Dong, J.F.; Schade, A.J.; Gardiner, E.E.; Kansas, G.S.; Li, C.Q.; McIntire, L.V.; Berndt, M.C.; Lopez, J.A. The glycoprotein ib-ix-v complex is a platelet counterreceptor for p-selectin. J. Exp. Med. 1999, 190, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin mac-1 (cd11b/cd18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sakuma, M.; Chen, Z.; Ustinov, V.; Shi, C.; Croce, K.; Zago, A.C.; Lopez, J.; Andre, P.; Plow, E.; et al. Leukocyte engagement of platelet glycoprotein ibalpha via the integrin mac-1 is critical for the biological response to vascular injury. Circulation 2005, 112, 2993–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Simon, D.I. Inflammation and thrombosis: The clot thickens. Circulation 2001, 103, 1718–1720. [Google Scholar] [CrossRef] [Green Version]
- Berndt, M.C.; Andrews, R.K. Bernard-soulier syndrome. Haematologica 2011, 96, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Pudas, R.; Heikkinen, O.; Permi, P.; Kilpelainen, I.; Munday, A.D.; Hartwig, J.H.; Stossel, T.P.; Ylanne, J. The structure of the gpib-filamin a complex. Blood 2006, 107, 1925–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaji, T.; Russell, S.; Ware, J. Amelioration of the macrothrombocytopenia associated with the murine bernard-soulier syndrome. Blood 2002, 100, 2102–2107. [Google Scholar] [CrossRef] [Green Version]
- Cranmer, S.L.; Pikovski, I.; Mangin, P.; Thompson, P.E.; Domagala, T.; Frazzetto, M.; Salem, H.H.; Jackson, S.P. Identification of a unique filamin a binding region within the cytoplasmic domain of glycoprotein ibalpha. Biochem. J. 2005, 387, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T. Qualitative disorders of platelets and megakaryocytes. J. Thromb. Haemost. 2005, 3, 1773–1782. [Google Scholar] [CrossRef]
- Lopez, J.A.; Andrews, R.K.; Afshar-Kharghan, V.; Berndt, M.C. Bernard-soulier syndrome. Blood 1998, 91, 4397–4418. [Google Scholar] [CrossRef]
- Bernard, J.; Soulier, J.P. On a new variety of congenital thrombocytary hemo-ragiparous dystrophy. Sem. Hop. 1948, 24, 3217–3223. [Google Scholar] [PubMed]
- Lanza, F. Bernard-soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J. Rare Dis. 2006, 1, 46. [Google Scholar] [CrossRef] [Green Version]
- Savoia, A.; Kunishima, S.; De Rocco, D.; Zieger, B.; Rand, M.L.; Pujol-Moix, N.; Caliskan, U.; Tokgoz, H.; Pecci, A.; Noris, P.; et al. Spectrum of the mutations in bernard-soulier syndrome. Hum. Mutat. 2014, 35, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lyle, V.A.; Cunningham, D. Mutation of leucine-57 to phenylalanine in a platelet glycoprotein ib alpha leucine tandem repeat occurring in patients with an autosomal dominant variant of bernard-soulier disease. Blood 1992, 79, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, M.L.; Diacovo, T.G.; Bainton, D.F.; Lanza, F.; Trejo, J.; Coughlin, S.R. Glycoprotein v-deficient platelets have undiminished thrombin responsiveness and do not exhibit a bernard-soulier phenotype. Blood 1999, 94, 4112–4121. [Google Scholar] [CrossRef]
- Weiss, H.J.; Tschopp, T.B.; Baumgartner, H.R.; Sussman, I.I.; Johnson, M.M.; Egan, J.J. Decreased adhesion of giant (bernard-soulier) platelets to subendothelium: Further implications on the role of the von willebrand factor in hemostasis. Am. J. Med. 1974, 57, 920–925. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Okumura, T. Reduced thrombin binding and aggregation in bernard-soulier platelets. J. Clin. Investig. 1978, 61, 861–864. [Google Scholar] [CrossRef]
- Dormann, D.; Clemetson, K.J.; Kehrel, B.E. The gpib thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood 2000, 96, 2469–2478. [Google Scholar] [CrossRef]
- Bevers, E.M.; Comfurius, P.; Nieuwenhuis, H.K.; Levy-Toledano, S.; Enouf, J.; Belluci, S.; Caen, J.P.; Zwaal, R.F. Platelet prothrombin converting activity in hereditary disorders of platelet function. Br. J. Haematol. 1986, 63, 335–345. [Google Scholar] [CrossRef]
- Pham, A.; Wang, J. Bernard-soulier syndrome: An inherited platelet disorder. Arch. Pathol. Lab. Med. 2007, 131, 1834–1836. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L.; Iolascon, A.; Savoia, A. Inherited thrombocytopenias: From genes to therapy. Haematologica 2002, 87, 860–880. [Google Scholar]
- Harrison, P.; Robinson, M.; Liesner, R.; Khair, K.; Cohen, H.; Mackie, I.; Machin, S. The pfa-100: A potential rapid screening tool for the assessment of platelet dysfunction. Clin. Lab. Haematol. 2002, 24, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Bolton-Maggs, P.H.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the ukhcdo. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Kosta, M.; Alessi, M.C.; Hezard, N. Laboratory techniques used to diagnose constitutional platelet dysfunction. Hämostaseologie 2020, 40, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Berndt, M.C. Bernard-soulier syndrome: An update. Semin. Thromb. Hemost. 2013, 39, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.J.; Sherman, G.G.; Glencross, D.K. Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of bernard-soulier syndrome. Pediatr. Hematol. Oncol. 1997, 14, 43–50. [Google Scholar] [CrossRef]
- Othman, M.; Emsley, J. Gene of the issue: Gp1ba gene mutations associated with bleeding. Platelets 2017, 28, 832–836. [Google Scholar] [CrossRef]
- Tait, A.S.; Cranmer, S.L.; Jackson, S.P.; Dawes, I.W.; Chong, B.H. Phenotype changes resulting in high-affinity binding of von willebrand factor to recombinant glycoprotein ib-ix: Analysis of the platelet-type von willebrand disease mutations. Blood 2001, 98, 1812–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Othman, M. Platelet-type von willebrand disease: Three decades in the life of a rare bleeding disorder. Blood Rev. 2011, 25, 147–153. [Google Scholar] [CrossRef]
- Franchini, M.; Montagnana, M.; Lippi, G. Clinical, laboratory and therapeutic aspects of platelet-type von willebrand disease. Int. J. Lab. Hematol. 2008, 30, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Boselli, B.D.; Kupinski, J.M. In vivo interaction of von willebrand factor with platelets following cryoprecipitate transfusion in platelet-type von willebrand’s disease. Blood 1984, 63, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, E.J. 2b or not 2b? What is the role of vwf in platelet-matrix interactions? And what is the role of the vwf:Cb in vwd diagnostics? These are the questions. J. Thromb. Haemost. 2006, 4, 892–894. [Google Scholar] [CrossRef]
- Othman, M.; Gresele, P. Guidance on the diagnosis and management of platelet-type von willebrand disease: A communication from the platelet physiology subcommittee of the isth. J. Thromb. Haemost. 2020, 18, 1855–1858. [Google Scholar] [CrossRef]
- Giannini, S.; Cecchetti, L.; Mezzasoma, A.M.; Gresele, P. Diagnosis of platelet-type von willebrand disease by flow cytometry. Haematologica 2010, 95, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [Green Version]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Research 2018, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.S.; Heijnen, H.F.; Raposo, G. Lysosome-related organelles: Unusual compartments become mainstream. Curr. Opin. Cell Biol. 2013, 25, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Heijnen, H.F.; Debili, N.; Vainchencker, W.; Breton-Gorius, J.; Geuze, H.J.; Sixma, J.J. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood 1998, 91, 2313–2325. [Google Scholar] [CrossRef]
- Youssefian, T.; Cramer, E.M. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood 2000, 95, 4004–4007. [Google Scholar] [CrossRef] [PubMed]
- Maynard, D.M.; Heijnen, H.F.; Horne, M.K.; White, J.G.; Gahl, W.A. Proteomic analysis of platelet alpha-granules using mass spectrometry. J. Thromb. Haemost. 2007, 5, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Flaumenhaft, R. Platelet secretion. In Platelets in Thrombotic and Non-Thrombotic Disorders; Gresele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 353–366. [Google Scholar]
- Youssefian, T.; Massé, J.-M.; Rendu, F.; Guichard, J.; Cramer, E.M. Platelet and megakaryocyte dense-granules contain glycoproteins ib and iib-iiia. Blood 1997, 89, 4047–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendu, F.; Brohard-Bohn, B. The platelet release reaction: Granules’ constituents, secretion and functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef]
- Weiss, H.J.; Witte, L.D.; Kaplan, K.L.; Lages, B.A.; Chernoff, A.; Nossel, H.L.; Goodman, D.S.; Baumgartner, H.R. Heterogeneity in storage pool deficiency: Studies on granule-bound substances in 18 patients including variants deficient in alpha-granules, platelet factor 4, beta-thromboglobulin, and platelet-derived growth factor. Blood 1979, 54, 1296–1319. [Google Scholar] [CrossRef] [Green Version]
- Mumford, A.D.; Frelinger, A.L., 3rd; Gachet, C.; Gresele, P.; Noris, P.; Harrison, P.; Mezzano, D. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb. Haemost. 2015, 114, 14–25. [Google Scholar] [CrossRef]
- Nurden, A.; Nurden, P. Advances in our understanding of the molecular basis of disorders of platelet function. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 76–91. [Google Scholar] [CrossRef] [PubMed]
- Sandrock, K.; Zieger, B. Current strategies in diagnosis of inherited storage pool defects. Transfus. Med. Hemother. 2010, 37, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules... or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Raccuglia, G. Gray platelet syndrome. A variety of qualitative platelet disorder. Am. J. Med. 1971, 51, 818–828. [Google Scholar] [CrossRef]
- Gerrard, J.M.; Phillips, D.R.; Rao, G.H.; Plow, E.F.; Walz, D.A.; Ross, R.; Harker, L.A.; White, J.G. Biochemical studies of two patients with the gray platelet syndrome. Selective deficiency of platelet alpha granules. J. Clin. Investig. 1980, 66, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T.; Nurden, P. The gray platelet syndrome: Clinical spectrum of the disease. Blood Rev. 2007, 21, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Jantunen, E.; Hanninen, A.; Naukkarinen, A.; Vornanen, M.; Lahtinen, R. Gray platelet syndrome with splenomegaly and signs of extramedullary hematopoiesis: A case report with review of the literature. Am. J. Hematol. 1994, 46, 218–224. [Google Scholar] [CrossRef]
- Caen, J.P.; Deschamps, J.F.; Bodevin, E.; Bryckaert, M.C.; Dupuy, E.; Wasteson, A. Megakaryocytes and myelofibrosis in gray platelet syndrome. Nouv. Rev. Fr. Hematol. 1987, 29, 109–114. [Google Scholar] [PubMed]
- Sims, M.C.; Mayer, L.; Collins, J.H.; Bariana, T.K.; Megy, K.; Lavenu-Bombled, C.; Seyres, D.; Kollipara, L.; Burden, F.S.; Greene, D.; et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood 2020, 136, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Breton-Gorius, J.; Vainchenker, W.; Nurden, A.; Levy-Toledano, S.; Caen, J. Defective alpha-granule production in megakaryocytes from gray platelet syndrome: Ultrastructural studies of bone marrow cells and megakaryocytes growing in culture from blood precursors. Am. J. Pathol. 1981, 102, 10–19. [Google Scholar] [PubMed]
- Simon, D.; Kunicki, T.; Nugent, D. Platelet function defects. Haemophilia 2008, 14, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.; Jasztal, M.; Pardo, M.; Aguera de Haro, S.; Collins, J.; Bariana, T.K.; Smethurst, P.A.; Grassi, L.; Petersen, R.; Nurden, P.; et al. Nbeal2 interacts with dock7, sec16a, and vac14. Blood 2018, 131, 1000–1011. [Google Scholar] [CrossRef]
- Lo, R.W.; Li, L.; Leung, R.; Pluthero, F.G.; Kahr, W.H.A. Nbeal2 (neurobeachin-like 2) is required for retention of cargo proteins by -granules during their production by megakaryocytes. Arter. Thromb. Vasc. Biol. 2018, 38, 2435–2447. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T.; Nurden, P. Should any genetic defect affecting alpha-granules in platelets be classified as gray platelet syndrome? Am. J. Hematol. 2016, 91, 714–718. [Google Scholar] [CrossRef]
- Villeneuve, J.; Block, A.; Le Bousse-Kerdiles, M.C.; Lepreux, S.; Nurden, P.; Ripoche, J.; Nurden, A.T. Tissue inhibitors of matrix metalloproteinases in platelets and megakaryocytes: A novel organization for these secreted proteins. Exp. Hematol. 2009, 37, 849–856. [Google Scholar] [CrossRef]
- Rosa, J.P.; George, J.N.; Bainton, D.F.; Nurden, A.T.; Caen, J.P.; McEver, R.P. Gray platelet syndrome. Demonstration of alpha granule membranes that can fuse with the cell surface. J. Clin. Investig. 1987, 80, 1138–1146. [Google Scholar] [CrossRef]
- Drouin, A.; Favier, R.; Masse, J.M.; Debili, N.; Schmitt, A.; Elbim, C.; Guichard, J.; Adam, M.; Gougerot-Pocidalo, M.A.; Cramer, E.M. Newly recognized cellular abnormalities in the gray platelet syndrome. Blood 2001, 98, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Lages, B.; Sussman, I.I.; Levine, S.P.; Coletti, D.; Weiss, H.J. Platelet alpha granule deficiency associated with decreased p-selectin and selective impairment of thrombin-induced activation in a new patient with gray platelet syndrome (alpha-storage pool deficiency). J. Lab. Clin. Med. 1997, 129, 364–375. [Google Scholar] [CrossRef]
- Shahraki, H.; Dorgalaleh, A.; Bain, B.J. Gray platelet syndrome (gps). In Congenital Bleeding Disorders: Diagnosis and Management; Dorgalaleh, A., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 379–396. [Google Scholar]
- Podda, G.; Femia, E.A.; Pugliano, M.; Cattaneo, M. Congenital defects of platelet function. Platelets 2012, 23, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Helip-Wooley, A.; Westbroek, W.; Gunay-Aygun, M.; Gahl, W.A. Disorders of lysosome-related organelle biogenesis: Clinical and molecular genetics. Annu Rev. Genom. Hum. Genet. 2008, 9, 359–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, A.; Bordet, J.C.; Eckly, A.; Gachet, C. Platelet delta-storage pool disease: An update. J. Clin. Med. 2020, 9, 2508. [Google Scholar] [CrossRef] [PubMed]
- Woldie, I.; Guo, R.; Ososki, R.; Dyson, G.; Mohamad, S.; Raval, K.K.; Gabali, A.M. Clinical Characteristics of Patients Diagnosed with Delta Granule Platelet Storage Pool Deficiency (Δ-PSPD); The Detroit Medical Center (DMC): Detroit, MI, USA, 2017. [Google Scholar]
- Masliah-Planchon, J.; Darnige, L.; Bellucci, S. Molecular determinants of platelet delta storage pool deficiencies: An update. Br. J. Haematol. 2013, 160, 5–11. [Google Scholar] [CrossRef]
- Nieuwenhuis, H.K.; Akkerman, J.W.; Sixma, J.J. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency: Studies on one hundred six patients. Blood 1987, 70, 620–623. [Google Scholar] [CrossRef] [Green Version]
- White, J.G. Use of the electron microscope for diagnosis of platelet disorders. Semin. Thromb. Hemost. 1998, 24, 163–168. [Google Scholar] [CrossRef]
- Wall, J.E.; Buijswilts, M.; Arnold, J.T.; Wang, W.; White, M.M.; Jennings, L.K.; Jackson, C.W. A flow cytometric assay using mepacrine for study of uptake and release of platelet dense granule contents. Br. J. Haematol. 1995, 89, 380–385. [Google Scholar] [CrossRef]
- Daskalakis, M.; Colucci, G.; Keller, P.; Rochat, S.; Silzle, T.; Biasiutti, F.D.; Barizzi, G.; Alberio, L. Decreased generation of procoagulant platelets detected by flow cytometric analysis in patients with bleeding diathesis. Cytometry B Clin. Cytom 2014, 86, 397–409. [Google Scholar] [CrossRef]
- Gordon, N.; Thom, J.; Cole, C.; Baker, R. Rapid detection of hereditary and acquired platelet storage pool deficiency by flow cytometry. Br. J. Haematol. 1995, 89, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Mullier, F.; Frotscher, B.; Briquel, M.E.; Toussaint, M.; Massin, F.; Lecompte, T.; Latger-Cannard, V. Usefulness of flow cytometric mepacrine uptake/release combined with cd63 assay in diagnosis of patients with suspected platelet dense granule disorder. Semin. Thromb. Hemost. 2016, 42, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Holmsen, H.; Weiss, H.J. Secretable storage pools in platelets. Annu. Rev. Med. 1979, 30, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin signaling: The platelet paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef] [Green Version]
- Poon, M.C.; Di Minno, G.; d’Oiron, R.; Zotz, R. New insights into the treatment of glanzmann thrombasthenia. Transfus. Med. Rev. 2016, 30, 92–99. [Google Scholar] [CrossRef] [Green Version]
- George, J.N.; Caen, J.P.; Nurden, A.T. Glanzmann’s thrombasthenia: The spectrum of clinical disease. Blood 1990, 75, 1383–1395. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, G.; Margaglione, M.; Glansmann’s Thrombasthemia Italian T. Glanzmann’s thrombasthenia: Modulation of clinical phenotype by alpha2c807t gene polymorphism. Haematologica 2003, 88, 1378–1382. [Google Scholar] [CrossRef]
- Di Minno, G.; Zotz, R.B.; d’Oiron, R.; Bindslev, N.; Di Minno, M.N.; Poon, M.C.; Glanzmann Thrombasthenia Registry Investigators. The international, prospective glanzmann thrombasthenia registry: Treatment modalities and outcomes of non-surgical bleeding episodes in patients with glanzmann thrombasthenia. Haematologica 2015, 100, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T. Glanzmann thrombasthenia. Orphanet J. Rare Dis. 2006, 1, 10. [Google Scholar] [CrossRef]
- Bellucci, S.; Caen, J. Molecular basis of glanzmann’s thrombasthenia and current strategies in treatment. Blood Rev. 2002, 16, 193–202. [Google Scholar] [CrossRef]
- Botero, J.P.; Lee, K.; Branchford, B.R.; Bray, P.F.; Freson, K.; Lambert, M.P.; Luo, M.; Mohan, S.; Ross, J.E.; Bergmeier, W.; et al. Glanzmann thrombasthenia: Genetic basis and clinical correlates. Haematologica 2020, 105, 888–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caen, J.P. Glanzmann’s thrombasthenia. Baillieres Clin. Haematol. 1989, 2, 609–625. [Google Scholar] [CrossRef]
- Linden, M.D.; Frelinger, A.L., 3rd; Barnard, M.R.; Przyklenk, K.; Furman, M.I.; Michelson, A.D. Application of flow cytometry to platelet disorders. Semin. Thromb. Hemost. 2004, 30, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Turitto, V.T.; Baumgartner, H.R. Further evidence that glycoprotein iib-iiia mediates platelet spreading on subendothelium. Thromb. Haemost. 1991, 65, 202–205. [Google Scholar] [CrossRef]
- Weiss, H.J.; Turitto, V.T.; Baumgartner, H.R. Platelet adhesion and thrombus formation on subendothelium in platelets deficient in glycoproteins iib-iiia, ib, and storage granules. Blood 1986, 67, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Jurk, K.; Kehrel, B.E. Inherited and acquired disorders of platelet function. Transfus. Med. Hemotherapy 2007, 34, 6–19. [Google Scholar] [CrossRef]
- Gobbi, G.; Sponzilli, I.; Mirandola, P.; Tazzari, P.L.; Caimi, L.; Cacchioli, A.; Matteucci, A.; Giuliani Piccari, G.; Cocco, L.; Vitale, M. Efficient platelet delta-granule release induced by [Ca2+]i elevation is modulated by gpiibiiia. Int. J. Mol. Med. 2006, 18, 309–313. [Google Scholar]
- Dorgalaleh, A.; Poon, M.; Shiravand, Y. Glanzmann thrombasthenia. In Congenital Bleeding Disorders; Dorgalaleh, A., Ed.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Gresele, P.; Subcommittee on Platelet Physiology of the International Society on T. Hemostasis. Diagnosis of inherited platelet function disorders: Guidance from the ssc of the isth. J. Thromb. Haemost. 2015, 13, 314–322. [Google Scholar] [CrossRef]
- Alessi, M.C.; Sie, P.; Payrastre, B. Strengths and weaknesses of light transmission aggregometry in diagnosing hereditary platelet function disorders. J. Clin. Med. 2020, 9, 763. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T.; Pillois, X.; Wilcox, D.A. Glanzmann thrombasthenia: State of the art and future directions. Semin Thromb. Hemost. 2013, 39, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, F.; Stierle, A.; Fournier, D.; Morales, M.; Andre, G.; Nurden, A.T.; Cazenave, J.P. A new variant of glanzmann’s thrombasthenia (strasbourg i). Platelets with functionally defective glycoprotein iib-iiia complexes and a glycoprotein iiia 214arg—214trp mutation. J. Clin. Investig. 1992, 89, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Nagata, S.; Sakuragi, T.; Segawa, K. Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 2020, 62, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Schoenwaelder, S.M. Procoagulant platelets: Are they necrotic? Blood 2010, 116, 2011–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kruchten, R.; Mattheij, N.J.; Saunders, C.; Feijge, M.A.; Swieringa, F.; Wolfs, J.L.; Collins, P.W.; Heemskerk, J.W.; Bevers, E.M. Both tmem16f-dependent and tmem16f-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 2013, 121, 1850–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by tmem16f. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. Tmem16f forms a ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Podoplelova, N.A.; Sveshnikova, A.N.; Kotova, Y.N.; Eckly, A.; Receveur, N.; Nechipurenko, D.Y.; Obydennyi, S.I.; Kireev, I.I.; Gachet, C.; Ataullakhanov, F.I.; et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood 2016, 128, 1745–1755. [Google Scholar] [CrossRef]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Lipid-protein interactions in blood coagulation. Biochim. Biophys. Acta 1998, 1376, 433–453. [Google Scholar] [CrossRef]
- Stenflo, J.; Fernlund, P.; Egan, W.; Roepstorff, P. Vitamin k dependent modifications of glutamic acid residues in prothrombin. Proc. Natl. Acad. Sci. USA 1974, 71, 2730–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, C. Gamma-carboxyglutamate-containing proteins and the vitamin k-dependent carboxylase. Biochem. J. 1990, 266, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, Y.Z.; Tajkhorshid, E. Distinct structural and adhesive roles of ca2+ in membrane binding of blood coagulation factors. Structure 2008, 16, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by gla domains of vitamin k-dependent proteins. Nat. Struct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef]
- Morel, O.; Toti, F.; Hugel, B.; Bakouboula, B.; Camoin-Jau, L.; Dignat-George, F.; Freyssinet, J.M. Procoagulant microparticles: Disrupting the vascular homeostasis equation? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Dale, G.L.; Remenyi, G.; Friese, P. Quantitation of microparticles released from coated-platelets. J. Thromb. Haemost. 2005, 3, 2081–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Alberio, L.; Safa, O.; Clemetson, K.J.; Esmon, C.T.; Dale, G.L. Surface expression and functional characterization of alpha-granule factor v in human platelets: Effects of ionophore a23187, thrombin, collagen, and convulxin. Blood 2000, 95, 1694–1702. [Google Scholar] [CrossRef]
- Shaw, A.W.; Pureza, V.S.; Sligar, S.G.; Morrissey, J.H. The local phospholipid environment modulates the activation of blood clotting. J. Biol. Chem. 2007, 282, 6556–6563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, E.C.; Rand, M.L. Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front. Cardiovasc. Med. 2020, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Poole, A.W. Procoagulant platelets: Generation, function, and therapeutic targeting in thrombosis. Blood 2017, 130, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberio, L.; Ravanat, C.; Hechler, B.; Mangin, P.H.; Lanza, F.; Gachet, C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of coat platelet formation. Thromb. Haemost. 2017, 117, 1101–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, G.L. Coated-platelets: An emerging component of the procoagulant response. J. Thromb. Haemost. 2005, 3, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.C.; Stoner, J.A.; Dale, G.L.; Rabadi, M.; Prodan, C.I. Higher coated-platelet levels in acute stroke are associated with lower cognitive scores at three months post infarction. J. Stroke Cerebrovasc. Dis. 2019, 28, 2398–2406. [Google Scholar] [CrossRef]
- Agbani, E.O.; van den Bosch, M.T.; Brown, E.; Williams, C.M.; Mattheij, N.J.; Cosemans, J.M.; Collins, P.W.; Heemskerk, J.W.; Hers, I.; Poole, A.W. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation 2015, 132, 1414–1424. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.; Jackson, S.P. Platelet factor xiii and calpain negatively regulate integrin alphaiibbeta3 adhesive function and thrombus growth. J. Biol. Chem. 2004, 279, 30697–30706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazepa, M.; Hoffman, M.; Monroe, D. Superactivated platelets: Thrombus regulators, thrombin generators, and potential clinical targets. Arterioscler Thromb. Vasc. Biol. 2013, 33, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Pecci, A.; Balduini, C.L. Desmopressin and super platelets. Blood 2014, 123, 1779–1780. [Google Scholar] [CrossRef] [Green Version]
- Storrie, B. A tip of the cap to procoagulant platelets. Blood 2016, 128, 1668–1669. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.W. Procoagulant ’Zombie’ Platelets. Available online: https://academy.isth.org/isth/2017/berlin/186727/johan.heemskerk.procoagulant.zombie.platelets.html (accessed on 25 January 2021).
- Hua, V.M.; Abeynaike, L.; Glaros, E.; Campbell, H.; Pasalic, L.; Hogg, P.J.; Chen, V.M. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 2015, 126, 2852–2862. [Google Scholar] [CrossRef] [PubMed]
- Obydennyy, S.I.; Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 2016, 14, 1867–1881. [Google Scholar] [CrossRef]
- Aliotta, A.; Bertaggia Calderara, D.; Zermatten, M.G.; Alberio, L. Sodium-calcium exchanger reverse mode sustains dichotomous ion fluxes required for procoagulant coat platelet formation. Thromb. Haemost. 2020, 121, 309–321. [Google Scholar] [CrossRef]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattheij, N.J.; Gilio, K.; van Kruchten, R.; Jobe, S.M.; Wieschhaus, A.J.; Chishti, A.H.; Collins, P.; Heemskerk, J.W.; Cosemans, J.M. Dual mechanism of integrin alphaiibbeta3 closure in procoagulant platelets. J. Biol. Chem. 2013, 288, 13325–13336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, F.S.; Marcinkiewicz, M.; Walsh, P.N. Par-1-stimulated factor ixa binding to a small platelet subpopulation requires a pronounced and sustained increase of cytoplasmic calcium. Biochemistry 2006, 45, 7289–7298. [Google Scholar] [CrossRef] [Green Version]
- Dale, G.L.; Friese, P.; Batar, P.; Hamilton, S.F.; Reed, G.L.; Jackson, K.W.; Clemetson, K.J.; Alberio, L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 2002, 415, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Joseph, P.M.; Vincent, A.S.; Dale, G.L. Coated-platelet levels are influenced by smoking, aspirin, and selective serotonin reuptake inhibitors. J. Thromb. Haemost. 2007, 5, 2149–2151. [Google Scholar] [CrossRef] [PubMed]
- Dale, G.L. Procoagulant platelets: Further details but many more questions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1596–1597. [Google Scholar] [CrossRef] [Green Version]
- Aliotta, A.; Krusi, M.; Bertaggia Calderara, D.; Zermatten, M.G.; Gomez, F.J.; Batista Mesquita Sauvage, A.P.; Alberio, L. Characterization of procoagulant coat platelets in patients with glanzmann thrombasthenia. Int. J. Mol. Sci. 2020, 21, 9515. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Vicic, W.J.; Lages, B.A.; Rogers, J. Isolated deficiency of platelet procoagulant activity. Am. J. Med. 1979, 67, 206–213. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim. Biophys. Acta 2004, 1636, 119–128. [Google Scholar] [CrossRef] [PubMed]
- van Geffen, J.P.; Swieringa, F.; Heemskerk, J.W. Platelets and coagulation in thrombus formation: Aberrations in the scott syndrome. Thromb. Res. 2016, 141 (Suppl. S2), S12–S16. [Google Scholar] [CrossRef]
- Adler, M.; Kaufmann, J.; Alberio, L.; Nagler, M. Diagnostic utility of the isth bleeding assessment tool in patients with suspected platelet function disorders. J. Thromb. Haemost. 2019, 17, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Vincent, A.S.; Padmanabhan, R.; Dale, G.L. Coated-platelet levels are low in patients with spontaneous intracerebral hemorrhage. Stroke 2009, 40, 2578–2580. [Google Scholar] [CrossRef] [Green Version]
- Prodan, C.I.; Vincent, A.S.; Dale, G.L. Coated platelet levels correlate with bleed volume in patients with spontaneous intracerebral hemorrhage. Stroke 2010, 41, 1301–1303. [Google Scholar] [CrossRef] [Green Version]
- Prodan, C.I.; Stoner, J.A.; Dale, G.L. Lower coated-platelet levels are associated with increased mortality after spontaneous intracerebral hemorrhage. Stroke 2015, 46, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Prodan, C.I.; Vincent, A.S.; Kirkpatrick, A.C.; Hoover, S.L.; Dale, G.L. Higher levels of coated-platelets are observed in patients with subarachnoid hemorrhage but lower levels are associated with increased mortality at 30 days. J. Neurol. Sci. 2013, 334, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Stoner, J.A.; Gordon, D.L.; Dale, G.L. Cerebral microbleeds in nonlacunar brain infarction are associated with lower coated-platelet levels. J. Stroke Cerebrovasc. Dis. 2014, 23, e325–e330. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Stoner, J.A.; Cowan, L.D.; Dale, G.L. Lower coated-platelet levels are associated with early hemorrhagic transformation in patients with non-lacunar brain infarction. J. Thromb. Haemost. 2010, 8, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Pethe, K.; Dale, G.L. Coated-platelet levels may explain some variability in clinical phenotypes observed with severe hemophilia. J. Thromb. Haemost. 2010, 8, 1140–1142. [Google Scholar] [CrossRef]
- Lastrapes, K.K.; Mohammed, B.M.; Mazepa, M.A.; Martin, E.J.; Barrett, J.C.; Massey, G.V.; Kuhn, J.G.; Nolte, M.E.; Hoffman, M.; Monroe, D.M.; et al. Coated platelets and severe haemophilia a bleeding phenotype: Is there a connection? Haemophilia 2016, 22, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, G.; Szasz, R.; Debreceni, I.B.; Szarvas, M.; Batar, P.; Nagy, B., Jr.; Kappelmayer, J.; Udvardy, M. Comparison of coated-platelet levels in patients with essential thrombocythemia with and without hydroxyurea treatment. Platelets 2013, 24, 486–492. [Google Scholar] [CrossRef]
- Prodan, C.I.; Joseph, P.M.; Vincent, A.S.; Dale, G.L. Coated-platelets in ischemic stroke: Differences between lacunar and cortical stroke. J. Thromb. Haemost. 2008, 6, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Vincent, A.S.; Dale, G.L. Coated-platelet levels are elevated in patients with transient ischemic attack. Transl. Res. 2011, 158, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Stoner, J.A.; Cowan, L.D.; Dale, G.L. Higher coated-platelet levels are associated with stroke recurrence following nonlacunar brain infarction. J. Cereb. Blood Flow Metab. 2013, 33, 287–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, A.C.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Coated-platelets predict stroke at 30 days following tia. Neurology 2017, 89, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.C.; Stoner, J.A.; Dale, G.L.; Prodan, C.I. Elevated coated-platelets in symptomatic large-artery stenosis patients are associated with early stroke recurrence. Platelets 2014, 25, 93–96. [Google Scholar] [CrossRef]
- Kirkpatrick, A.C.; Tafur, A.J.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Coated-platelets improve prediction of stroke and transient ischemic attack in asymptomatic internal carotid artery stenosis. Stroke 2014, 45, 2995–3001. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, A.C.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Increased platelet procoagulant potential predicts recurrent stroke and tia after lacunar infarction. J. Thromb. Haemost. 2020, 18, 660–668. [Google Scholar] [CrossRef]
- Wang, L.; Bi, Y.; Yu, M.; Li, T.; Tong, D.; Yang, X.; Zhang, C.; Guo, L.; Wang, C.; Kou, Y.; et al. Phosphatidylserine-exposing blood cells and microparticles induce procoagulant activity in non-valvular atrial fibrillation. Int. J. Cardiol. 2018, 258, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Pasalic, L.; Wing-Lun, E.; Lau, J.K.; Campbell, H.; Pennings, G.J.; Lau, E.; Connor, D.; Liang, H.P.; Muller, D.; Kritharides, L.; et al. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J. Thromb. Haemost. 2018, 16, 1198–1210. [Google Scholar] [CrossRef]
- Kou, Y.; Zou, L.; Liu, R.; Zhao, X.; Wang, Y.; Zhang, C.; Dong, Z.; Kou, J.; Bi, Y.; Fu, L.; et al. Intravascular cells and circulating microparticles induce procoagulant activity via phosphatidylserine exposure in heart failure. J. Thromb. Thrombolysis 2019, 48, 187–194. [Google Scholar] [CrossRef]
- Ray, B.; Pandav, V.M.; Mathews, E.A.; Thompson, D.M.; Ford, L.; Yearout, L.K.; Bohnstedt, B.N.; Chaudhary, S.; Dale, G.L.; Prodan, C.I. Coated-platelet trends predict short-term clinical outcomeafter subarachnoid hemorrhage. Transl. Stroke Res. 2017, 9, 459–470. [Google Scholar] [CrossRef]
- Ray, B.; Ross, S.R.; Danala, G.; Aghaei, F.; Nouh, C.D.; Ford, L.; Hollabaugh, K.M.; Karfonta, B.N.; Santucci, J.A.; Cornwell, B.O.; et al. Systemic response of coated-platelet and peripheral blood inflammatory cell indices after aneurysmal subarachnoid hemorrhage and long-term clinical outcome. J. Crit. Care 2019, 52, 1–9. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Gosmanova, A.K.; Lyons, T.J.; May, K.D.; Dashti, A.; Baker, M.Z.; Olansky, L.; Aston, C.E.; Dale, G.L. Coated-platelet levels in patients with type 1 and with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 81, e8–e10. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.C.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Clopidogrel use and smoking cessation result in lower coated-platelet levels after stroke. Platelets 2019, 31, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, P.; Gillespie, S.; Armstrong, P.C.; Allan, H.E.; Warner, T.D.; Brohi, K. Histone h4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc. Natl. Acad. Sci. USA 2019, 116, 17444–17449. [Google Scholar] [CrossRef] [Green Version]
- Prodan, C.I.; Szasz, R.; Vincent, A.S.; Ross, E.D.; Dale, G.L. Coated-platelets retain amyloid precursor protein on their surface. Platelets 2006, 17, 56–60. [Google Scholar] [CrossRef]
- Prodan, C.I.; Ross, E.D.; Vincent, A.S.; Dale, G.L. Coated-platelets correlate with disease progression in alzheimer disease. J. Neurol. 2007, 254, 548–549. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Ross, E.D.; Vincent, A.S.; Dale, G.L. Rate of progression in alzheimer’s disease correlates with coated-platelet levels--a longitudinal study. Transl. Res. 2008, 152, 99–102. [Google Scholar] [CrossRef]
- Prodan, C.I.; Ross, E.D.; Vincent, A.S.; Dale, G.L. Coated-platelets are higher in amnestic versus nonamnestic patients with mild cognitive impairment. Alzheimer Dis. Assoc. Disord. 2007, 21, 259–261. [Google Scholar] [CrossRef]
- Prodan, C.I.; Ross, E.D.; Stoner, J.A.; Cowan, L.D.; Vincent, A.S.; Dale, G.L. Coated-platelet levels and progression from mild cognitive impairment to alzheimer disease. Neurology 2011, 76, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valaydon, Z.S.; Lee, P.; Dale, G.L.; Januszewski, A.S.; Rowley, K.G.; Nandurkar, H.; Karschimkus, C.; Best, J.D.; Lyons, T.J.; Jenkins, A.J. Increased coated-platelet levels in chronic haemodialysis patients. Nephrology 2009, 14, 148–154. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross talk pathways between coagulation and inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef]
- Kulkarni, S.; Woollard, K.J.; Thomas, S.; Oxley, D.; Jackson, S.P. Conversion of platelets from a proaggregatory to a proinflammatory adhesive phenotype: Role of paf in spatially regulating neutrophil adhesion and spreading. Blood 2007, 110, 1879–1886. [Google Scholar] [CrossRef] [Green Version]
- Charania, R.; Smith, J.; Vesely, S.K.; Dale, G.L.; Holter, J. Quantitation of coated platelet potential during collection, storage, and transfusion of apheresis platelets. Transfusion 2011, 51, 2690–2694. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia Calderara, D.; Crettaz, D.; Aliotta, A.; Barelli, S.; Tissot, J.D.; Prudent, M.; Alberio, L. Generation of procoagulant collagen- and thrombin-activated platelets in platelet concentrates derived from buffy coat: The role of processing, pathogen inactivation, and storage. Transfusion 2018, 58, 2395–2406. [Google Scholar] [CrossRef]
- Gerber, B.; Alberio, L.; Rochat, S.; Stenner, F.; Manz, M.G.; Buser, A.; Schanz, U.; Stussi, G. Safety and efficacy of cryopreserved autologous platelet concentrates in hla-alloimmunized patients with hematologic malignancies. Transfusion 2016, 56, 2426–2437. [Google Scholar] [CrossRef]
- Kotova, Y.N.; Ataullakhanov, F.I.; Panteleev, M.A. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5’diphosphate acting via the p2y12 receptor. J. Thromb. Haemost. 2008, 6, 1603–1605. [Google Scholar] [CrossRef]
- Norgard, N.B.; Saya, S.; Hann, C.L.; Hennebry, T.A.; Schechter, E.; Dale, G.L. Clopidogrel attenuates coated-platelet production in patients undergoing elective coronary catheterization. J. Cardiovasc. Pharmacol. 2008, 52, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Norgard, N.B.; Hann, C.L.; Dale, G.L. Cangrelor attenuates coated-platelet formation. Clin. Appl. Thromb. Hemost. 2009, 15, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, H.M.; Buckland, R.J.; Sugidachi, A.; Jakubowski, J.A.; Storey, R.F. The active metabolite of prasugrel effectively blocks the platelet p2y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets 2008, 19, 125–133. [Google Scholar] [CrossRef]
- Hamilton, S.F.; Miller, M.W.; Thompson, C.A.; Dale, G.L. Glycoprotein iib/iiia inhibitors increase coat-platelet production in vitro. J. Lab. Clin. Med. 2004, 143, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Vermylen, J.; Hoylaerts, M.; Arnout, J. Increased mortality with long-term platelet glycoprotein iib/iiia antagonists: An explanation? Circulation 2001, 104, E109. [Google Scholar] [CrossRef] [PubMed]
- Topalov, N.N.; Kotova, Y.N.; Vasil’ev, S.A.; Panteleev, M.A. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 2012, 157, 105–115. [Google Scholar] [CrossRef]
- van der Meijden, P.E.; Feijge, M.A.; Swieringa, F.; Gilio, K.; Nergiz-Unal, R.; Hamulyak, K.; Heemskerk, J.W. Key role of integrin alpha(iib)beta (3) signaling to syk kinase in tissue factor-induced thrombin generation. Cell. Mol. Life Sci. 2012, 69, 3481–3492. [Google Scholar] [CrossRef] [Green Version]
- Razmara, M.; Hu, H.; Masquelier, M.; Li, N. Glycoprotein iib/iiia blockade inhibits platelet aminophospholipid exposure by potentiating translocase and attenuating scramblase activity. Cell. Mol. Life Sci. 2007, 64, 999–1008. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Ruggeri, Z.M.; Pareti, F.I.; Capitanio, A. 1-deamino-8-d-arginine vasopressin: A new pharmacological approach to the management of haemophilia and von willebrands’ diseases. Lancet 1977, 1, 869–872. [Google Scholar] [CrossRef]
- Tomasiak, M.M.; Stelmach, H.; Bodzenta-Lukaszyk, A.; Tomasiak, M. Involvement of na+/h+ exchanger in desmopressin-induced platelet procoagulant response. Acta Biochim. Pol. 2004, 51, 773–788. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Ciborowski, M.; Radziwon, P. Vasopressin acts on platelets to generate procoagulant activity. Blood Coagul. Fibrinolysis 2008, 19, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Colucci, G.; Stutz, M.; Rochat, S.; Conte, T.; Pavicic, M.; Reusser, M.; Giabbani, E.; Huynh, A.; Thurlemann, C.; Keller, P.; et al. The effect of desmopressin on platelet function: A selective enhancement of procoagulant coat platelets in patients with primary platelet function defects. Blood 2014, 123, 1905–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swieringa, F.; Lance, M.D.; Fuchs, B.; Feijge, M.A.; Solecka, B.A.; Verheijen, L.P.; Hughes, K.R.; van Oerle, R.; Deckmyn, H.; Kannicht, C.; et al. Desmopressin treatment improves platelet function under flow in patients with postoperative bleeding. J. Thromb. Haemost. 2015, 13, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.T. Auranofin, a thioredoxin reductase inhibitor, causes platelet death through calcium overload. Platelets 2019, 30, 98–104. [Google Scholar] [CrossRef]
- Tseng, Y.L.; Braun, A.; Chang, J.P.; Chiang, M.L.; Tseng, C.Y.; Chen, W. Micromolar concentrations of citalopram or escitalopram inhibit glycoprotein vi-mediated and integrin alphaiibbeta3-mediated signaling in human platelets. Toxicol. Appl. Pharmacol. 2019, 364, 106–113. [Google Scholar] [CrossRef]
- Galan, A.M.; Lopez-Vilchez, I.; Diaz-Ricart, M.; Navalon, F.; Gomez, E.; Gasto, C.; Escolar, G. Serotonergic mechanisms enhance platelet-mediated thrombogenicity. Thromb. Haemost. 2009, 102, 511–519. [Google Scholar] [CrossRef]
- Laporte, S.; Chapelle, C.; Caillet, P.; Beyens, M.N.; Bellet, F.; Delavenne, X.; Mismetti, P.; Bertoletti, L. Bleeding risk under selective serotonin reuptake inhibitor (ssri) antidepressants: A meta-analysis of observational studies. Pharmacol. Res. 2017, 118, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Mezei, G.; Debreceni, I.B.; Kerenyi, A.; Remenyi, G.; Szasz, R.; Illes, A.; Kappelmayer, J.; Batar, P. Dasatinib inhibits coated-platelet generation in patients with chronic myeloid leukemia. Platelets 2019, 30, 836–843. [Google Scholar] [CrossRef]
- Deb, S.; Boknas, N.; Sjostrom, C.; Tharmakulanathan, A.; Lotfi, K.; Ramstrom, S. Varying effects of tyrosine kinase inhibitors on platelet function-a need for individualized cml treatment to minimize the risk for hemostatic and thrombotic complications? Cancer Med. 2020, 9, 313–323. [Google Scholar] [CrossRef]
- Tullemans, B.M.E.; Nagy, M.; Sabrkhany, S.; Griffioen, A.W.; Oude Egbrink, M.G.A.; Aarts, M.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Tyrosine kinase inhibitor pazopanib inhibits platelet procoagulant activity in renal cell carcinoma patients. Front. Cardiovasc. Med. 2018, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Umbach, A.T.; Bissinger, R.; Gawaz, M.; Lang, F. Inhibition of collagen related peptide induced platelet activation and apoptosis by ceritinib. Cell Physiol. Biochem. 2018, 45, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Keuren, J.F.; Wielders, S.J.; Ulrichts, H.; Hackeng, T.; Heemskerk, J.W.; Deckmyn, H.; Bevers, E.M.; Lindhout, T. Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Williams, C.M.; Hers, I.; Poole, A.W. Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci. Rep. 2017, 7, 2770. [Google Scholar] [CrossRef] [Green Version]
- Denorme, F.; Manne, B.K.; Portier, I.; Eustes, A.S.; Kosaka, Y.; Kile, B.T.; Rondina, M.T.; Campbell, R.A. Platelet necrosis mediates ischemic stroke outcome in mice. Blood 2020, 135, 429–440. [Google Scholar] [CrossRef]
- Choo, H.J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Sodergren, A.L.; Ramstrom, S. Platelet subpopulations remain despite strong dual agonist stimulation and can be characterised using a novel six-colour flow cytometry protocol. Sci. Rep. 2018, 8, 1441. [Google Scholar] [CrossRef] [Green Version]
- Topalov, N.N.; Yakimenko, A.O.; Canault, M.; Artemenko, E.O.; Zakharova, N.V.; Abaeva, A.A.; Loosveld, M.; Ataullakhanov, F.I.; Nurden, A.T.; Alessi, M.C.; et al. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin alpha(iib)beta(3). Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Szasz, R.; Dale, G.L. Coat platelets. Curr. Opin. Hematol. 2003, 10, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Abaeva, A.A.; Canault, M.; Kotova, Y.N.; Obydennyy, S.I.; Yakimenko, A.O.; Podoplelova, N.A.; Kolyadko, V.N.; Chambost, H.; Mazurov, A.V.; Ataullakhanov, F.I.; et al. Procoagulant platelets form an alpha-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J. Biol. Chem. 2013, 288, 29621–29632. [Google Scholar] [CrossRef] [Green Version]
- Aupeix, K.; Hugel, B.; Martin, T.; Bischoff, P.; Lill, H.; Pasquali, J.L.; Freyssinet, J.M. The significance of shed membrane particles during programmed cell death in vitro, and in vivo, in hiv-1 infection. J. Clin. Investig. 1997, 99, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Bohling, S.D.; Pagano, M.B.; Stitzel, M.R.; Ferrell, C.; Yeung, W.; Chandler, W.L. Comparison of clot-based vs chromogenic factor xa procoagulant phospholipid activity assays. Am. J. Clin. Pathol. 2012, 137, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemker, H.C.; Wielders, S.; Kessels, H.; Beguin, S. Continuous registration of thrombin generation in plasma, its use for the determination of the thrombin potential. Thromb. Haemost. 1993, 70, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Zermatten, M.G.; Fraga, M.; Calderara, D.B.; Aliotta, A.; Moradpour, D.; Alberio, L. Biomarkers of liver dysfunction correlate with a prothrombotic and not with a prohaemorrhagic profile in patients with cirrhosis. JHEP Rep. Innov. Hepatol. 2020, 2, 100120. [Google Scholar] [CrossRef] [PubMed]
- Camire, R.M.; Kalafatis, M.; Simioni, P.; Girolami, A.; Tracy, P.B. Platelet-derived factor va/va leiden cofactor activities are sustained on the surface of activated platelets despite the presence of activated protein c. Blood 1998, 91, 2818–2829. [Google Scholar] [CrossRef] [Green Version]
- Thuerlemann, C.; Haeberli, A.; Alberio, L. Monitoring thrombin generation by electrochemistry: Development of an amperometric biosensor screening test for plasma and whole blood. Clin. Chem. 2009, 55, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Jy, W.; Horstman, L.L.; Jimenez, J.J.; Ahn, Y.S.; Biro, E.; Nieuwland, R.; Sturk, A.; Dignat-George, F.; Sabatier, F.; Camoin-Jau, L.; et al. Measuring circulating cell-derived microparticles. J. Thromb. Haemost. 2004, 2, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Robert, S.; Poncelet, P.; Dignat-George, F. Overcoming limitations of microparticle measurement by flow cytometry. Semin. Thromb. Hemost. 2010, 36, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.; Beguin, S.; Andree, H.; Hemker, H.C. Measurement of thrombin generation in whole blood--the effect of heparin and aspirin. Thromb. Haemost. 1994, 72, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Ninivaggi, M.; Apitz-Castro, R.; Dargaud, Y.; de Laat, B.; Hemker, H.C.; Lindhout, T. Whole-blood thrombin generation monitored with a calibrated automated thrombogram-based assay. Clin. Chem. 2012, 58, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Prior, S.M.; Mann, K.G.; Freeman, K.; Butenas, S. Continuous thrombin generation in whole blood: New applications for assessing activators and inhibitors of coagulation. Anal. Biochem. 2018, 551, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Konings, J.; Yan, Q.; Kelchtermans, H.; Kremers, R.; de Laat, B.; Roest, M. A novel assay for studying the involvement of blood cells in whole blood thrombin generation. J. Thromb. Haemost. 2020, 18, 1291–1301. [Google Scholar] [CrossRef] [Green Version]
- Hemker, H.C.; Giesen, P.L.; Ramjee, M.; Wagenvoord, R.; Beguin, S. The thrombogram: Monitoring thrombin generation in platelet-rich plasma. Thromb. Haemost. 2000, 83, 589–591. [Google Scholar] [CrossRef] [Green Version]
- Douxfils, J.; Morimont, L.; Bouvy, C.; de Saint-Hubert, M.; Devalet, B.; Devroye, C.; Dincq, A.S.; Dogne, J.M.; Guldenpfennig, M.; Baudar, J.; et al. Assessment of the analytical performances and sample stability on st genesia system using the stg-drugscreen application. J. Thromb. Haemost. 2019, 17, 1273–1287. [Google Scholar] [CrossRef]
- Talon, L.; Sinegre, T.; Lecompte, T.; Pereira, B.; Massoulie, S.; Abergel, A.; Lebreton, A. Hypercoagulability (thrombin generation) in patients with cirrhosis is detected with st-genesia. J. Thromb. Haemost. 2020, 18, 2177–2190. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia Calderara, D.; Zermatten, M.G.; Aliotta, A.; Batista Mesquita Sauvage, A.P.; Carle, V.; Heinis, C.; Alberio, L. Tissue factor-independent coagulation correlates with clinical phenotype in factor xi deficiency and replacement therapy. Thromb. Haemost. 2021, 121, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, E.M.; Kuprash, A.D.; Dashkevich, N.M.; Vardanyan, D.M.; Chernyakov, A.V.; Kumskova, M.A.; Nair, S.C.; Srivastava, A.; Ataullakhanov, F.I.; Panteleev, M.A.; et al. Determination of fibrin clot growth and spatial thrombin propagation in the presence of different types of phospholipid surfaces. Platelets 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Aswad, M.H.; Kissova, J.; Rihova, L.; Zavrelova, J.; Ovesna, P.; Penka, M. High level of circulating microparticles in patients with bcr/abl negative myeloproliferative neoplasm—A pilot study. Klein. Onkol. 2019, 32, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, M.J.; Bradford, R.; Hobl, E.L.; Lin, F.C.; Jilma, B.; Key, N.S. Procoagulant microparticles promote coagulation in a factor xi-dependent manner in human endotoxemia. J. Thromb. Haemost. 2016, 14, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Exner, T.; Joseph, J.E.; Connor, D.; Low, J.; Ma, D.D. Increased procoagulant phospholipid activity in blood from patients with suspected acute coronary syndromes: A pilot study. Blood Coagul. Fibrinolysis 2005, 16, 375–379. [Google Scholar] [CrossRef]
- Marchetti, M.; Tartari, C.J.; Russo, L.; Panova-Noeva, M.; Leuzzi, A.; Rambaldi, A.; Finazzi, G.; Woodhams, B.; Falanga, A. Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia. Am. J. Hematol. 2014, 89, 68–73. [Google Scholar] [CrossRef] [Green Version]
- van Dreden, P.; Rousseau, A.; Fontaine, S.; Woodhams, B.J.; Exner, T. Clinical evaluation of a new functional test for detection of plasma procoagulant phospholipids. Blood Coagul. Fibrinolysis 2009, 20, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.K.; Abdel-Monem, H.; Niravath, P.; Le, A.; Bellera, R.V.; Langlois, K.; Nagata, S.; Rumbaut, R.E.; Thiagarajan, P. Lactadherin and clearance of platelet-derived microvesicles. Blood 2009, 113, 1332–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, D.; Talar, M.; Watala, C.; Przygodzki, T. Intravital assessment of blood platelet function. A review of the methodological approaches with examples of studies of selected aspects of blood platelet function. Int. J. Mol. Sci. 2020, 21, 8334. [Google Scholar] [CrossRef] [PubMed]
- Montague, S.J.; Lim, Y.J.; Lee, W.M.; Gardiner, E.E. Imaging platelet processes and function-current and emerging approaches for imaging in vitro and in vivo. Front. Immunol. 2020, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Marcinczyk, N.; Golaszewska, A.; Misztal, T.; Gromotowicz-Poplawska, A.; Rusak, T.; Chabielska, E. New approaches for the assessment of platelet activation status in thrombus under flow condition using confocal microscopy. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. Tmem16f is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef] [Green Version]
- Nechipurenko, D.Y.; Receveur, N.; Yakimenko, A.O.; Shepelyuk, T.O.; Yakusheva, A.A.; Kerimov, R.R.; Obydennyy, S.I.; Eckly, A.; Leon, C.; Gachet, C.; et al. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arter. Thromb. Vasc. Biol. 2019, 39, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kennedy, D.R.; Lin, L.; Huang, M.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of beta3 integrins. Blood 2012, 120, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, C.; Methia, N.; Frenette, P.S.; Rayburn, H.; Ullman-Cullere, M.; Hynes, R.O.; Wagner, D.D. A mouse model of severe von willebrand disease: Defects in hemostasis and thrombosis. Proc. Natl. Acad. Sci. USA 1998, 95, 9524–9529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deppermann, C.; Cherpokova, D.; Nurden, P.; Schulz, J.N.; Thielmann, I.; Kraft, P.; Vogtle, T.; Kleinschnitz, C.; Dutting, S.; Krohne, G.; et al. Gray platelet syndrome and defective thrombo-inflammation in nbeal2-deficient mice. J. Clin. Investig. 2013, 123, 3331–3342. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe sars-cov-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Gasecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic complications in patients with covid-19: Pathophysiological mechanisms, diagnosis, and treatment. Cardiovasc. Drugs Ther. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to covid-19 thromboinflammation. Nat. Rev. Cardiol. 2020, 1–16. [Google Scholar] [CrossRef]
- Larsen, J.B.; Pasalic, L.; Hvas, A.M. Platelets in coronavirus disease 2019. Semin. Thromb. Hemost. 2020, 46, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Freedman, J.E. Platelets and covid-19: Inflammation, hyperactivation and additional questions. Circ. Res. 2020, 127, 1419–1421. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M. Potential role of platelets in covid-19: Implications for thrombosis. Res. Pract. Thromb. Haemost. 2020, 4, 737–740. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. Sars-cov-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death. Dis. 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets can associate with sars-cov-2 rna and are hyperactivated in covid-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with covid-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe covid-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Manne, B.K.; Portier, I.; Petrey, A.C.; Middleton, E.A.; Kile, B.T.; Rondina, M.T.; Campbell, R.A. Covid-19 patients exhibit reduced procoagulant platelet responses. J. Thromb. Haemost. 2020, 18, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in covid-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Maclay, J.D.; McAllister, D.A.; Johnston, S.; Raftis, J.; McGuinnes, C.; Deans, A.; Newby, D.E.; Mills, N.L.; MacNee, W. Increased platelet activation in patients with stable and acute exacerbation of copd. Thorax 2011, 66, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, T.; Mairbaurl, H.; Pleisch, B.; Maggiorini, M.; Bartsch, P.; Reinhart, W.H. Platelet count and function at high altitude and in high-altitude pulmonary edema. J. Appl. Physiol. 2006, 100, 690–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, T.; Ahmad, S.; Gupta, N.; Sahu, A.; Ahmad, Y.; Nair, V.; Chatterjee, T.; Bajaj, N.; Sengupta, S.; Ganju, L.; et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood 2014, 123, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.L.; Russell, J.; Granger, D.N. Platelet activation and platelet-leukocyte aggregation elicited in experimental colitis are mediated by interleukin-6. Inflamm. Bowel Dis. 2014, 20, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Senchenkova, E.Y.; Komoto, S.; Russell, J.; Almeida-Paula, L.D.; Yan, L.S.; Zhang, S.; Granger, D.N. Interleukin-6 mediates the platelet abnormalities and thrombogenesis associated with experimental colitis. Am. J. Pathol. 2013, 183, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, S.A.; Peng, J.; Friese, P.; Wolf, R.F.; Harrison, P.; Downs, T.; Hamilton, K.; Comp, P.; Dale, G.L. Cytokine-induced alteration of platelet and hemostatic function. Stem Cells 1996, 14 (Suppl. S1), 154–162. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. Ros in platelet biology: Functional aspects and methodological insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Endpoint | Description | Common Markers | Phenotype in Procoagulant Platelets | Phenotype in Non-Procoagulant Platelets |
|---|---|---|---|---|
| Necrotic-like | ||||
| Phosphatidylserine | Negatively charged amino-phospholipids of platelet membrane bilayer, contribute to the procoagulant activity | Annexin V, lactadherin | Positive | Negative |
| Mitochondrial membrane depolarization | Mitochondrial events (depolarization) are implicated in platelet procoagulant activity process | Rhodamine (such as TMRM) | Low TMRM staining | High TMRM staining |
| JC-1 | Lower JC-1 fluorescence ratio (red/green) | Higher JC-1 fluorescence ratio (red/green) | ||
| Fibrinogen receptor GPIIb/IIIa (integrin αIIbβ3) | Platelet membrane glycoprotein; in its activated conformation binds to fibrinogen and mediates platelet aggregation | Anti-CD41/CD61 IgM antibody recognizing the activated conformation (PAC-1) | Negative | Positive |
| Platelet surface coating by α-granule proteins | Proteins present in α-granule secreted upon platelet activation and retained on the platelet surface by a serotonin- and transglutaminase mechanism | Specific antibodies against α-granule proteins, such as FV/Va, fibrinogen, VWF, fibronectin, thrombospondin, and α2-antiplasmin | Positive | Negative |
| Type of Sample | Assay What Does It Measure? | Assay Name and Principle | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| WB | Coagulation potential (subsampling TG measurement) | TGA chromogenic | Presence of all blood cells and coagulation factors | Tedious subsampling at interval points; Time consuming; Only a snapshot picture of TG is available | [228] |
| Coagulation potential (continuous TG measurement) | TGA Paper based WB-TG assay Fluorogenic (rhodamine 110-based thrombin substrate) | Close to physiological haemostasis; Presence of all blood cells and coagulation factors | Potential of procoagulant platelets is not specifically targeted; Calibration is difficult because of haemolysis and/or haematocrit might vary in WB sample; Interference of contact activation; Needs experienced operator | [229,230] | |
| TGA Novel WB-TG assay Fluorogenic (rhodamine 110-based thrombin substrate) | Close to physiological haemostasis; Presence of all blood cells and coagulation factors; Stable light transmission achieved by continuous mixing of the assay plate | Potential of procoagulant platelets is not specifically targeted; | [231] | ||
| PRP | Coagulation potential (continuous TG measurement) | TGA e.g., Thrombinoscope (Stago), Techno-thrombin (Techno-clone) Fluorogenic | Mimics in vivo condition; Consider the interaction of platelets and coagulation factors | Potential of procoagulant platelets is not specifically targeted; Standardization is difficult; Reactivity of platelets: easy to provoke unwanted activation | [232] |
| PPP | Coagulation potential (continuous TG measurement) | TGA e.g., Thrombinoscope (Stago), ST Genesia (Stago) Fluorogenic | Defined concentration of tissue factor and artificial phospholipids; Standardization possible in automated version; Possible to store frozen samples | Potential of procoagulant platelets is not specifically targeted; Do not consider the interaction of platelets with coagulation factors; Loss of sensitivity for the intrinsic pathway if high amount of TF is used | [222,233] |
| TM-TGA ST Genesia (Stago), Fluorogenic | To study the role of protein C system by comparison of TM− and TM+ samples | TGA automated version: exact tissue factor concentration is not communicated | [223,234] | ||
| Spatio-temporal dynamics of coagulation (real time TG and fibrin clot formation) | Thrombodynamics Video microscopy system based on measurements of light scattering images intensity | Pre-analytics is standardized; TG and fibrin formation measured at the same time; Allows to investigate separately TF-dependent and TF-independent coagulation; PRP can be added to the mix | Problematic with lipemic samples; Available only in specialized laboratory | [235,236] | |
| Gel filtered or washed platelets | Coagulation potential (continuous TG) | Modified TGA assay fluorogenic | Targets specific procoagulant populations | Preparation is laborious; Requires experienced operator | [126,201] |
| Quantifies the number of procoagulant platelets | Flow cytometry fluorescence | Targets procoagulant platelet formation and associated markers | [131] | ||
| Measures the rate of clot growth | Experimental video microscopy Based on intensity of light scattering images | Specifically assess the contribution of activated platelets to clot growth | Requires experienced operator | [126] | |
| PMPs | Quantifies procoagulant potential of PMPs expressing PS. | Zymuphen MP Activity assay (Hyphen BioMed) ELISA, chromogenic | Easy to perform; High speed of sample analysis | Size of the PMPs can affect binding to Annexin V, thus lower detection of PS; No information on count, size or origin | [220,221,237,238] |
| Procoagulant potential of PMPs expressing PS added to phospholipid free plasma | Procoag PPL (Stago) Clotting time Number of PMPs is inversely proportional to clotting time | Can be used also on WB, PRP, PPP; Easy to perform | No information on count, size or origin | [239,240,241] | |
| Quantifies PMPs derived from gel filtered/washed platelets | Flow cytometry fluorescence Identification of PMPs by size (FSC) and fluorescence (e.g., bodily-label) | Target PMPs derived specifically from procoagulant platelets; Gel filtration/washing remove plasmatic components | PMPs are close to electronic noise and debris, part of the population might be below the threshold Require expertise and sensitive cytometer | [125,242] | |
| Coagulation potential (continuous TG) | Modified TGA Fluorogenic Isolation of PMPs by centrifugation | Specifically assess contribution of PMPs derived from procoagulant platelets to TG | Preparation is laborious | [126] | |
| Measures the rate of clot growth | Experimental video microscopy systemBased on intensity of light scattering images | Specifically assess the contribution of PMPs isolated from activated platelets to clot growth | Require experienced operator | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliotta, A.; Bertaggia Calderara, D.; Zermatten, M.G.; Marchetti, M.; Alberio, L. Thrombocytopathies: Not Just Aggregation Defects—The Clinical Relevance of Procoagulant Platelets. J. Clin. Med. 2021, 10, 894. https://doi.org/10.3390/jcm10050894
Aliotta A, Bertaggia Calderara D, Zermatten MG, Marchetti M, Alberio L. Thrombocytopathies: Not Just Aggregation Defects—The Clinical Relevance of Procoagulant Platelets. Journal of Clinical Medicine. 2021; 10(5):894. https://doi.org/10.3390/jcm10050894
Chicago/Turabian StyleAliotta, Alessandro, Debora Bertaggia Calderara, Maxime G. Zermatten, Matteo Marchetti, and Lorenzo Alberio. 2021. "Thrombocytopathies: Not Just Aggregation Defects—The Clinical Relevance of Procoagulant Platelets" Journal of Clinical Medicine 10, no. 5: 894. https://doi.org/10.3390/jcm10050894
APA StyleAliotta, A., Bertaggia Calderara, D., Zermatten, M. G., Marchetti, M., & Alberio, L. (2021). Thrombocytopathies: Not Just Aggregation Defects—The Clinical Relevance of Procoagulant Platelets. Journal of Clinical Medicine, 10(5), 894. https://doi.org/10.3390/jcm10050894