
Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2


{kind=link}
{kind=link}
Abstract
:1. Introduction
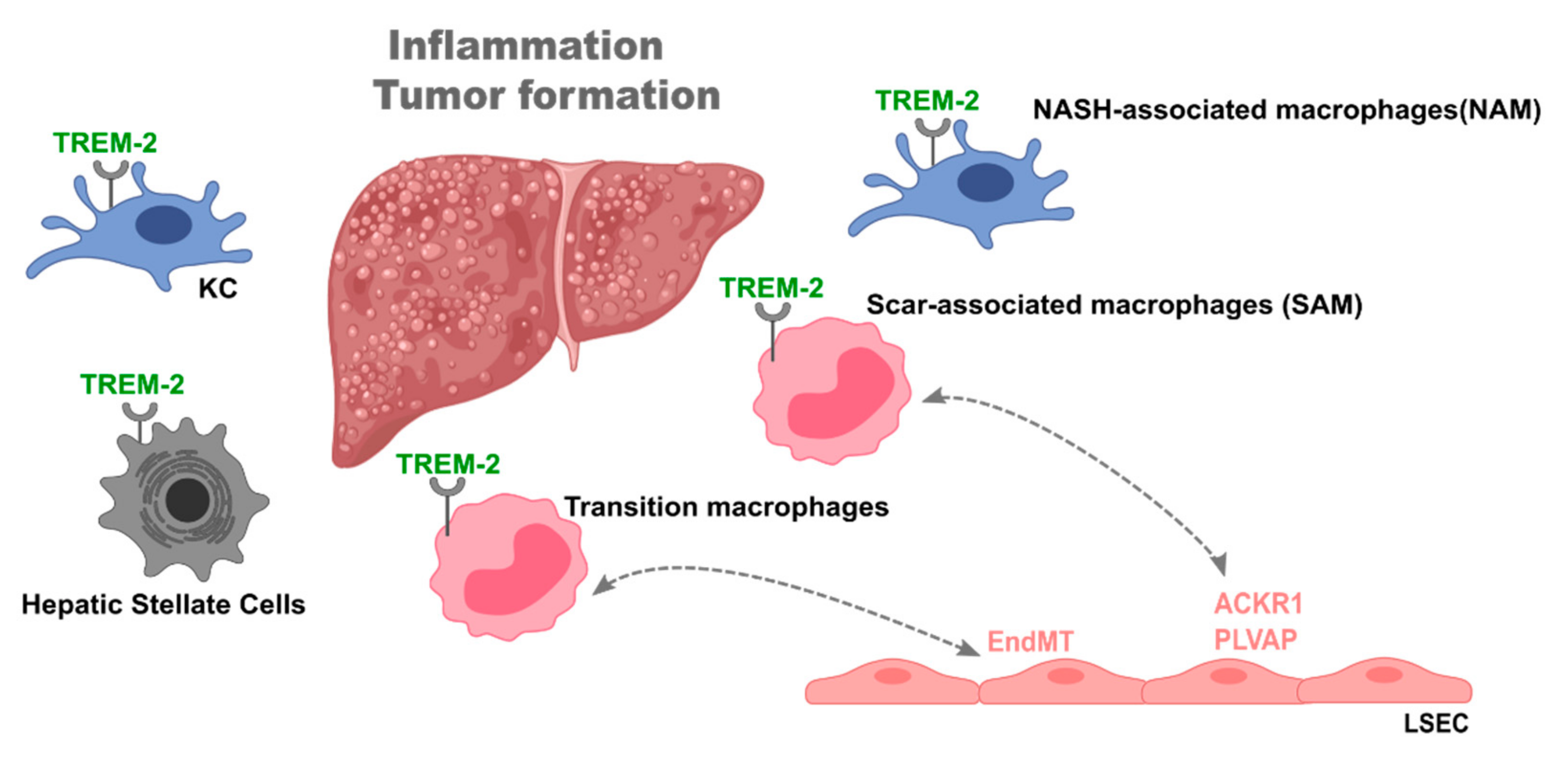
2. Macrophages in the Liver
2.1. Liver Resident Macrophages—Kupffer Cells
2.2. Macrophages in Liver Disease
2.2.1. Macrophages and Non-Alcoholic Fatty Liver Disease (NAFLD)
2.2.2. Macrophages and Chronic Liver Disease
3. Liver Endothelial Cells
3.1. Liver Sinusoidal Endothelial Cells (LSECs)
3.2. Liver Sinusoidal Endothelial Cells (LSECs) and Liver Disease
4. Macrophages and Endothelial Cells Crosstalk
4.1. Macrophages and Endothelial Cells in Homeostasis
4.2. Macrophages and Endothelial Cells in Liver Disease
4.3. Trem-2 as a Likely Mediator of Macrophage/Monocyte-Endothelial Cell Interactions
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Seo, W. Hepatic non-parenchymal cells: Master regulators of alcoholic liver disease? World J. Gastroenterol. 2016, 22, 1348. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, D.E.; Johnson, K.; Ryan, L.; Boorman, G.; Maronpot, R.R. New insights into functional aspects of liver morphology. Toxicol. Pathol. 2005, 33, 27–34. [Google Scholar] [CrossRef]
- Duarte, N.; Coelho, I.C.; Patarrão, R.S.; Almeida, J.I.; Penha-Gonçalves, C.; Macedo, M.P. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed Res. Int. 2015, 2015, 984578. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef]
- Furth, R.V.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 45, 845–852. [Google Scholar] [CrossRef]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 1–18. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Chen, J.-B.; Tsai, T.-F.; Tsai, Y.-C.; Tsai, C.-Y.; Liang, P.-H.; Hsu, T.-L.; Wu, C.-Y.; Netea, M.G.; Wong, C.-H.; et al. CLEC4F Is an Inducible C-Type Lectin in F4/80-Positive Cells and Is Involved in Alpha-Galactosylceramide Presentation in Liver. PLoS ONE 2013, 8, e65070. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Gordon, S.; Pl, A. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [Green Version]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”—How macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflügers Arch. Eur. J. Physiol. 2017, 469, 397–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Guilliams, M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018, 69, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Devisscher, L.; Verhelst, X.; Colle, I.; Van Vlierberghe, H.; Geerts, A. The role of macrophages in obesity-driven chronic liver disease. J. Leukoc. Biol. 2016, 99, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.J.; Leon, P.; Ryan, J.C. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology 2008, 48, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Lanthier, N.; Molendi-Coste, O.; Horsmans, Y.; van Rooijen, N.; Cani, P.D.; Leclercq, I.A. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Liver Physiol. 2010, 298, G107–G116. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with Vitamin, E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; Van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; Doherty, R.M.O. Depletion of Liver Kupffer Cells Prevents the Development of Diet-Induced Hepatic Steatosis and Insulin Resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C Chemokine Receptor 2 (CCR2) Regulates the Hepatic Recruitment of Myeloid Cells That Promote Obesity-Induced Hepatic Steatosis. Diabetes 2010, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.B.; Liu, Y.; Liu, C.; Xiang, X.; Wang, J.; Cheng, Z.; Shah, S.V.; Zhang, S.; Zhang, L.; Zhuang, X.; et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009, 50, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Ramachandran, P.; Iredale, J.P. Reversibility of liver fibrosis. Ann. Hepatol. 2009, 8, 283–291. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J. A Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Sanyal, A.; Vlad, R.; Harrison, S.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. Cenicriviroc versus Placebo for the Treatment of Nonalcoholic Steatohepatitis with Liver Fibrosis: Results from the Year 1 Primary Analysis of the Phase 2b. Hepatology 2015, 64, 1118–1119. [Google Scholar]
- Ding, B.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, T.; Naito, H.; Suehiro, J.-i.; Lin, Y.; Kawaji, H.; Iba, T.; Kouno, T.; Ishikawa-Kato, S.; Furuno, M.; Takara, K.; et al. CD157 Marks Tissue-Resident Endothelial Stem Cells with Homeostatic and Regenerative Properties. Cell Stem Cell 2018, 22, 384–397.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.; Maretti-Mira, A. Liver Sinusoidal Endothelial Cell: An Update. Semin. Liver Dis. 2017, 37, 377–387. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, L.; Chiu, J.D.; Van De Ven, G.; Gaarde, W.A.; Deleve, L.D. Hepatic vascular endothelial growth factor regulates recruitment of rat liver sinusoidal endothelial cell progenitor cells. Gastroenterology 2012, 143, 1555–1563.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harb, R.; Xie, G.; Lutzko, C.; Guo, Y.; Wang, X.; Hill, C.K.; Kanel, G.C.; DeLeve, L.D. Bone Marrow Progenitor Cells Repair Rat Hepatic Sinusoidal Endothelial Cells after Liver Injury. Gastroenterology 2009, 137, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Wisse, F.; Braet, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 12, 1–17. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Sun, X.; Harris, E.N. New aspects of hepatic endothelial cells in physiology and nonalcoholic fatty liver disease. Am. J. Physiol. Physiol. 2020, 318, C1200–C1213. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD / NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Zhang, Q.; Xiao, W.; Shao, M.; Fan, Q.; Zhang, H.; Zou, Y.; Li, X.; Xu, W.; Mo, Z.; et al. Protective effects of Sapindus mukorossi Gaertn against fatty liver disease induced by high fat diet in rats. Biochem. Biophys. Res. Commun. 2014, 450, 685–691. [Google Scholar] [CrossRef]
- Hammoutene, A.; Rautou, P. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefere, S.; Van de Velde, F.; Hoorens, A.; Raevens, S.; Van Campenhout, S.; Vandierendonck, A.; Neyt, S.; Vandeghinste, B.; Vanhove, C.; Debbaut, C.; et al. Angiopoietin-2 Promotes Pathological Angiogenesis and Is a Therapeutic Target in Murine Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1087–1104. [Google Scholar] [CrossRef]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyachi, Y.; Tsuchiya, K.; Komiya, C.; Shiba, K.; Shimazu, N.; Yamaguchi, S.; Deushi, M.; Osaka, M.; Inoue, K.; Sato, Y.; et al. Roles for Cell-Cell Adhesion and Contact in Obesity-Induced Hepatic Myeloid Cell Accumulation and Article Roles for Cell-Cell Adhesion and Contact in Obesity-Induced Hepatic Myeloid Cell Accumulation and Glucose Intolerance. Cell Rep. 2017, 18, 2766–2779. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef]
- Wu, L.Q.; Zhang, W.J.; Niu, J.X.; Ye, L.Y.; Yang, Z.H.; Grau, G.E.; Lou, J.N. Phenotypic and Functional Differences between Human Liver Cancer Endothelial Cells and Liver Sinusoidal Endothelial Cells. J. Vasc. Res. 2008, 45, 78–86. [Google Scholar] [CrossRef]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribera, J.; Pauta, M.; Melgar-lesmes, P.; Córdoba, B.; Bosch, A.; Calvo, M.; Rodrigo-torres, D.; Sancho-bru, P.; Mira, A.; Jiménez, W.; et al. A small population of liver endothelial cells undergoes endothelial-to- mesenchymal transition in response to chronic liver injury. Liver Biliary Tract Physiol. Pathophysiol. 2017, 492–504. [Google Scholar] [CrossRef]
- Lin, F.; Wang, N.; Zhang, T. The Role of Endothelial–Mesenchymal Transition in Development and Pathological Process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef]
- Medici, D. Endothelial-Mesenchymal Transition in Regenerative Medicine. Stem Cells Int. 2016, 2016, 6962801. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory Cytokines Promote Mesenchymal Transformation in Embryonic and Adult Valve Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.-S.; Seki, E. Chemokines and Chemokine Receptors in the Development of NAFLD. In Obesity, Fatty Liver and Liver Cancer; Springer: Berlin/Heidelberg, Germany, 2018; pp. 45–53. [Google Scholar]
- Kalucka, J.; Bierhansl, L.; Wielockx, B.; Carmeliet, P.; Eelen, G. Interaction of endothelial cells with macrophages—Linking molecular and metabolic signaling. Pflugers Arch. Eur. J. Physiol. 2017, 469, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgar-Lesmes, P.; Edelman, E.R. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 2015, 63, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalor, P.F.; Tuncer, C.; Weston, C.; Martin-Santos, A.; Smith, D.J.; Adams, D.H. Vascular Adhesion Protein-1 as a Potential Therapeutic Target in Liver Disease. Ann. N. Y. Acad. Sci. 2007, 1110, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, B.; Menger, M.D. The Hepatic Microcirculation: Mechanistic Contributions and Therapeutic Targets in Liver Injury and Repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef] [PubMed]
- Spolarics, Z. Endotoxemia, pentose cycle, and the oxidant/antioxidant balance in the hepatic sinusoid. J. Leukoc. Biol. 1998, 63, 534–541. [Google Scholar] [CrossRef]
- Schubert, S.Y.; Benarroch, A.; Monter-Solans, J.; Edelman, E.R. Primary Monocytes Regulate Endothelial Cell Survival through Secretion of Angiopoietin-1 and Activation of Endothelial Tie2. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Schubert, S.Y.; Benarroch, A.; Ostvang, J.; Edelman, E.R. Regulation of Endothelial Cell Proliferation by Primary Monocytes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Antonov, A.S.; Munn, D.H.; Kolodgie, F.D.; Virmani, R.; Gerrity, R.G. Aortic endothelial cells regulate proliferation of human monocytes in vitro via a mechanism synergistic with macrophage colony-stimulating factor. J. Clin. Investig. 1997, 99, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhou, Y.; Wang, H.; Zhang, M.; Qiu, P.; Zhang, M.; Zhang, R.; Zhao, Q.; Liu, J. Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front. Immunol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff, S. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. 2019, 33, 2278–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.Q.; Muratore, C.S.; So, E.; Sun, C.; Dubielecka, P.M.; Reginato, A.M. M1 Macrophage e Induced Endothelial-to-Mesenchymal Transition Promotes Infantile Hemangioma Regression. Am. J. Pathol. 2017, 187, 2102–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M. Trems in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef]
- Kober, D.L.; Brett, T.J. TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 2017, 429, 1607–1629. [Google Scholar] [CrossRef]
- Park, M.; Yi, J.W.; Kim, E.M.; Yoon, I.J.; Lee, E.H.; Lee, H.Y.; Ji, K.Y.; Lee, K.H.; Jang, J.H.; Oh, S.S.; et al. Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes 2015, 64, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Perugorria, M.J.; Esparza-Baquer, A.; Oakley, F.; Labiano, I.; Korosec, A.; Jais, A.; Mann, J.; Tiniakos, D.; Santos-Laso, A.; Arbelaiz, A.; et al. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut 2019, 68, 533–546. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Koike, M.; Spusta, S.; Niemi, E.; Yenari, M.; Nakamura, C.M.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2011, 109, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Esparza-Baquer, A.; Labiano, I.; Sharif, O.; Agirre-Lizaso, A.; Oakley, F.; Rodrigues, P.M.; Zhuravleva, E.; O’Rourke, C.J.; Hijona, E.; Jimenez-Agüero, R.; et al. TREM-2 defends the liver against hepatocellular carcinoma through multifactorial protective mechanisms. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wang, Q.; Wang, S.; Zhang, J.; Liu, T.; Guo, L.; Yu, Y.; Lin, J.D. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol. Metab. 2019, 20, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level; Springer: New York, NY, USA, 2019; Volume 575, ISBN 4158601916. [Google Scholar]
- Coelho, I.; Duarte, N.; Barros, A.; Macedo, M.P.; Penha-Gonçalves, C. Trem-2 promotes emergence of restorative macrophages and endothelial cells during recovery from hepatic tissue damage. Front. Immunol. 2021. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, I.; Duarte, N.; Macedo, M.P.; Penha-Gonçalves, C. Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2. J. Clin. Med. 2021, 10, 1248. https://doi.org/10.3390/jcm10061248
Coelho I, Duarte N, Macedo MP, Penha-Gonçalves C. Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2. Journal of Clinical Medicine. 2021; 10(6):1248. https://doi.org/10.3390/jcm10061248
Chicago/Turabian StyleCoelho, Inês, Nádia Duarte, Maria Paula Macedo, and Carlos Penha-Gonçalves. 2021. "Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2" Journal of Clinical Medicine 10, no. 6: 1248. https://doi.org/10.3390/jcm10061248
APA StyleCoelho, I., Duarte, N., Macedo, M. P., & Penha-Gonçalves, C. (2021). Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2. Journal of Clinical Medicine, 10(6), 1248. https://doi.org/10.3390/jcm10061248