
Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Fluorescein Isothiocyanate (FITC)-Dextran Assay
2.3. Evans Blue Assay
2.4. ZO-1 and iNOS Immunostaining
2.5. Brain Glutathione (GSH) Levels
2.6. Determination of Brain Lipid Peroxidation by Measurement of Thiobarbituric acid Reactive Substances
2.7. Nitrite Determination
2.8. Statistical Analysis
3. Results
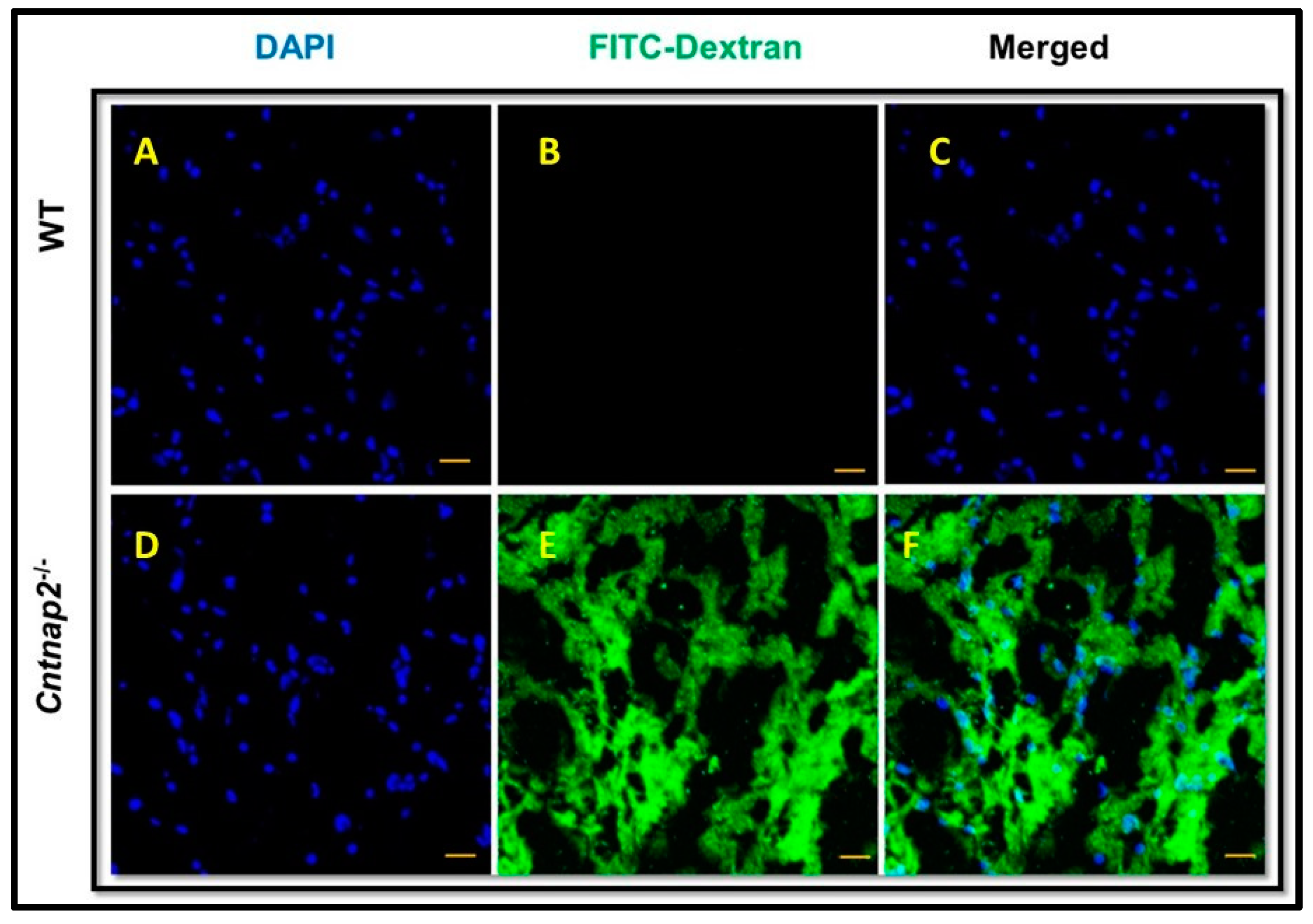
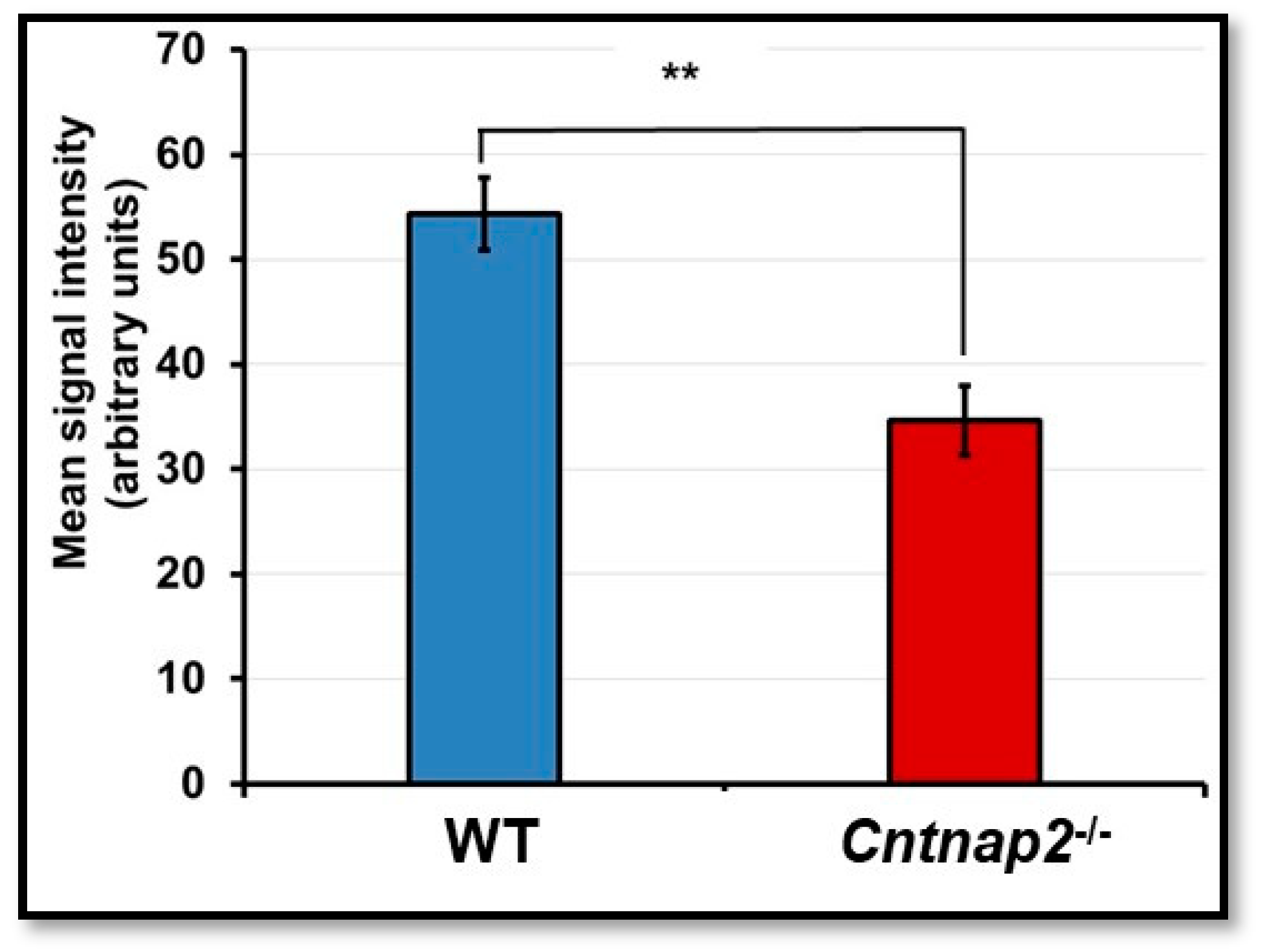
3.1. FITC-Dextran Assay
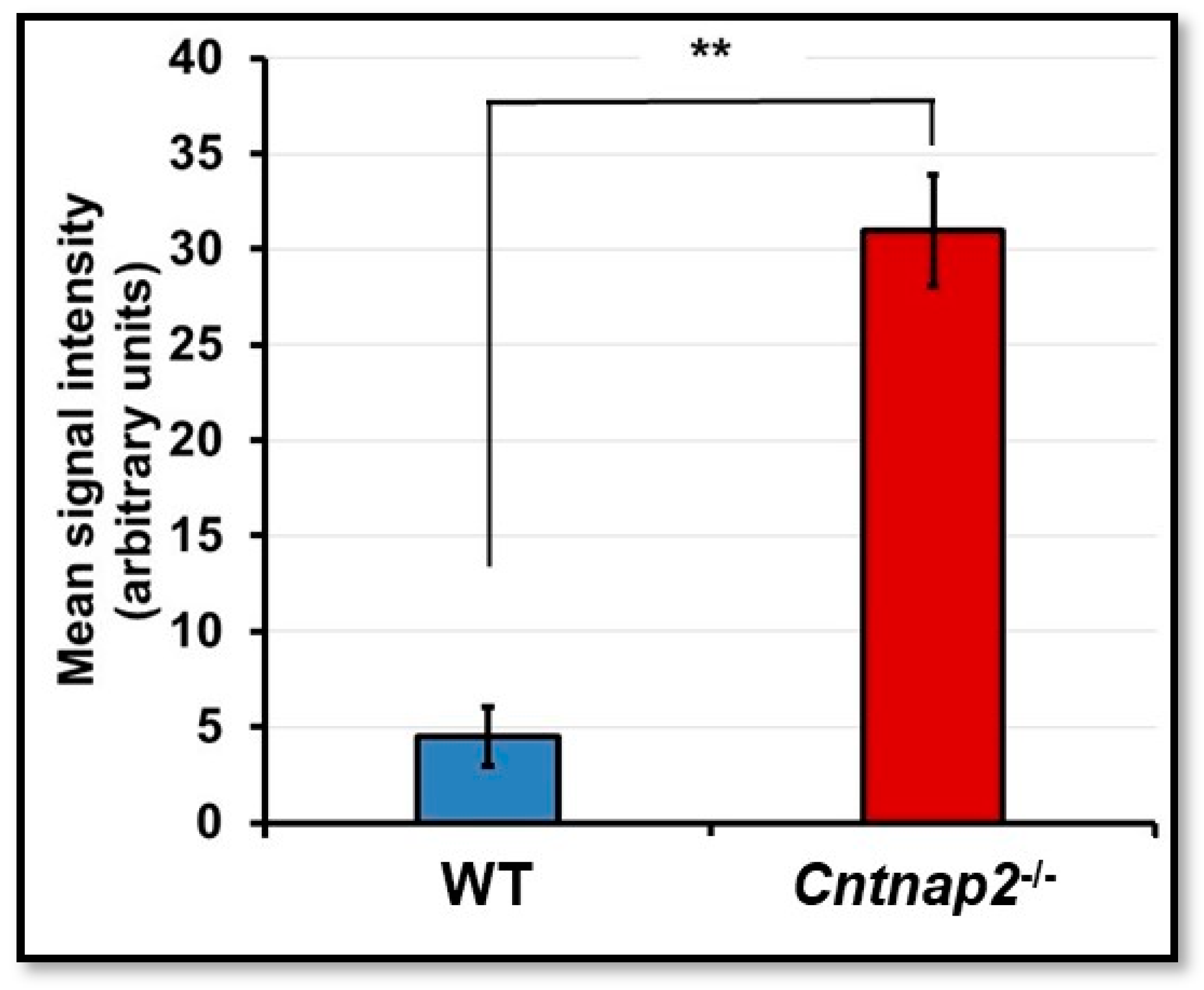
3.2. BBB Permeability Using Evans Blue Assay
3.3. ZO-1 Immunostaining
3.4. Oxidative Stress
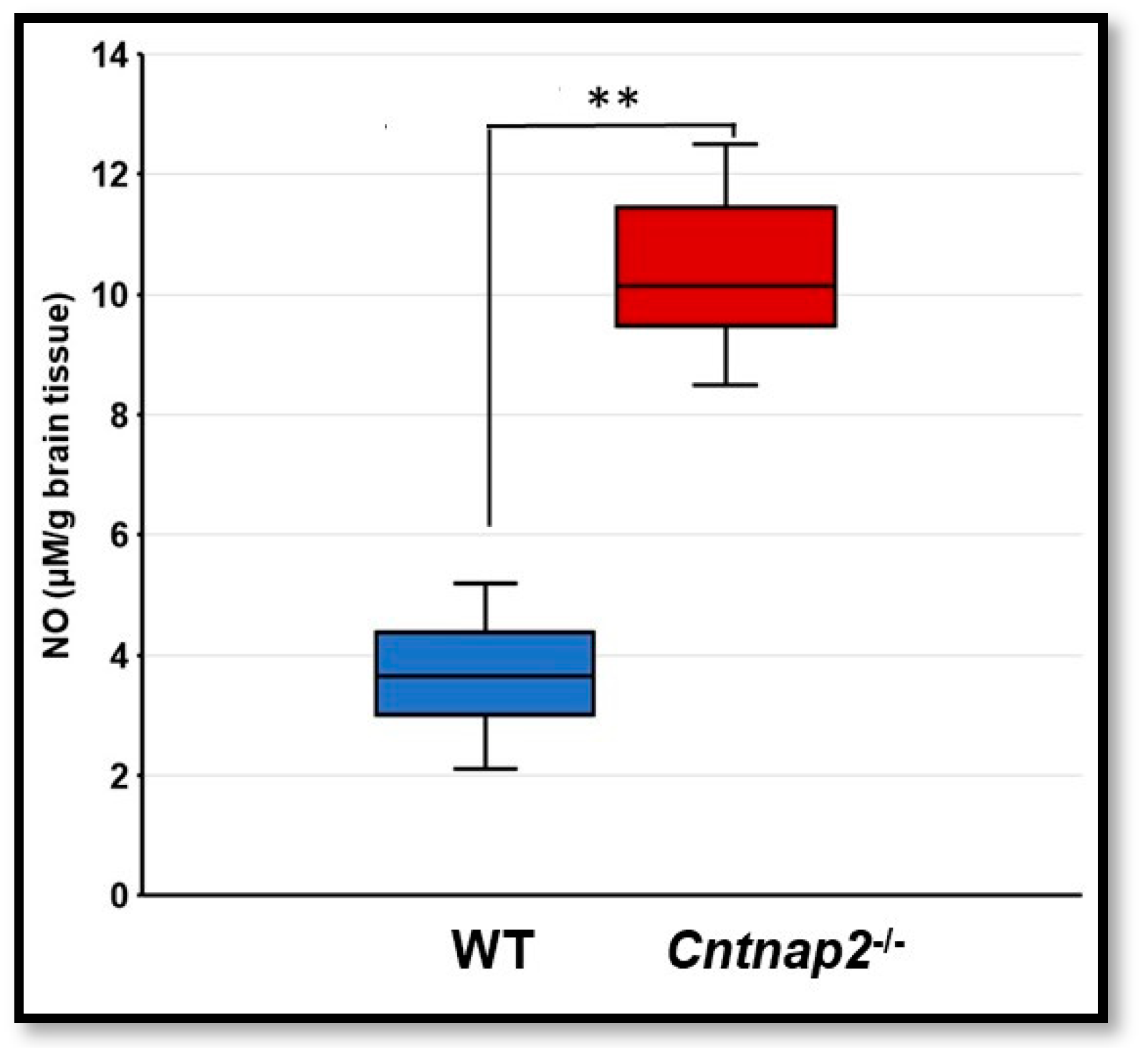
3.5. Nitrosative Stress in Cntnap2−/− Rats
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, R.P.; Milan, S.; Naigles, L.; Robins, D.L.; Barton, M.L.; Adamson, L.B.; Fein, D.A. Symptoms of autism spectrum disorder and developmental delay in children with low mental age. Clin. Neuropsychol. 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, united states, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Whitely, A.; Shandley, K.; Huynh, M.; Brown, C.M.; Austin, D.W.; Bhowmik, J. Brief report: Pregnancy, birth and infant feeding practices: A survey-based investigation into risk factors for autism spectrum disorder. J. Autism Dev. Disord. 2021. [Google Scholar] [CrossRef]
- Ijomone, O.M.; Olung, N.F.; Akingbade, G.T.; Okoh, C.O.A.; Aschner, M. Environmental influence on neurodevelopmental disorders: Potential association of heavy metal exposure and autism. J. Trace Elem. Med. Biol. 2020, 62, 126638. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative stress and immune system dysfunction in autism spectrum disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Liu, G.; Kay, S.-I.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and autism spectrum disorder: Is there a correlation? Front. Cell. Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Menezo, Y.J.R.; Elder, K.; Dale, B. Link between increased prevalence of autism spectrum disorder syndromes and oxidative stress, DNA methylation, and imprinting. JAMA Pediatr. 2015, 169, 1066. [Google Scholar] [CrossRef] [Green Version]
- Brault, V.; Nguyen, T.L.; Flores-Gutiérrez, J.; Iacono, G.; Birling, M.-C.; Lalanne, V.; Meziane, H.; Manousopoulou, A.; Pavlovic, G.; Lindner, L.; et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and down syndrome. PLoS Genet. 2021, 17, e1009777. [Google Scholar] [CrossRef]
- Bukatova, S.; Renczes, E.; Reichova, A.; Filo, J.; Sadlonova, A.; Mravec, B.; Ostatnikova, D.; Bakos, J.; Bacova, Z. Shank3 deficiency is associated with altered profile of neurotransmission markers in pups and adult mice. Neurochem. Res. 2021, 46, 3342–3355. [Google Scholar] [CrossRef] [PubMed]
- Bryda, E.C. The mighty mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar] [PubMed]
- Agarwala, S.; Ramachandra, N.B. Role of CNTNAP2 in autism manifestation outlines the regulation of signaling between neurons at the synapse. Egypt J. Med. Hum. Genet. 2021, 22, 22. [Google Scholar] [CrossRef]
- Fang, F.; Ge, M.; Liu, J.; Zhang, Z.; Yu, H.; Zhu, S.; Xu, L.; Shao, L. Association between Genetic Variants in DUSP15, CNTNAP2, and PCDHA Genes and Risk of Childhood Autism Spectrum Disorder. Behav. Neurol. 2021, 2021, 4150926. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Azima, A.; Aziz, M.A.; Aka, T.D.; Jafrin, S.; Millat, M.S.; Siddiqui, S.A.; Uddin, M.G.; Hussain, M.S.; Islam, M.S. CNTNAP2 gene polymorphisms in autism spectrum disorder and language impairment among Bangladeshi children: A case-control study combined with a meta-analysis. Hum. Cell. 2021, 34, 1410–1423. [Google Scholar] [CrossRef]
- Zare, S.; Mashayekhi, F.; Bidabadi, E. The association of CNTNAP2 Rs7794745 gene polymorphism and autism in Iranian population. J. Clin. Neurosci. 2017, 39, 189–192. [Google Scholar] [CrossRef]
- Rodenas-Cuadrado, P.; Pietrafusa, N.; Francavilla, T.; La Neve, A.; Striano, P.; Vernes, S.C. Characterisation of CASPR2 deficiency disorder—A syndrome involving autism, epilepsy and language impairment. BMC Med. Genet. 2016, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Chiocchetti, A.G.; Kopp, M.; Waltes, R.; Haslinger, D.; Duketis, E.; Jarczok, T.A.; Poustka, F.; Voran, A.; Graab, U.; Meyer, J.; et al. Variants of the CNTNAP2 5′ Promoter as risk factors for autism spectrum disorders: A genetic and functional approach. Mol. Psychiatry 2014, 20, 839–849. [Google Scholar] [CrossRef]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Hum. Genet. 2013, 22, 171–178. [Google Scholar] [CrossRef]
- Li, X.; Hu, Z.; He, Y.; Xiong, Z.; Long, Z.; Peng, Y.; Bu, F.; Ling, J.; Xun, G.; Mo, X.; et al. Association analysis of CNTNAP2 polymorphisms with autism in the Chinese Han population. Psychiatr. Genet. 2010, 20, 113–117. [Google Scholar] [CrossRef]
- Alarcón, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poot, M. Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders. Mol. Syndromol. 2015, 6, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñagarikano, O.; Geschwind, D.H. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol. Med. 2012, 18, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Martin, M.; Joubert, B.; Pellier-Monnin, V.; Pascual, O.; Noraz, N.; Honnorat, J. Contactin-Associated Protein-like 2, a Protein of the Neurexin Family Involved in Several Human Diseases. Eur. J. Neurosci. 2018, 48, 1906–1923. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Pierce, K.D.; Shaw, A.D.; Heath, A.; Mitchell, P.B.; Schofield, P.R.; Fullerton, J.M. Comprehensive cross-disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet. 2018, 14, e1007535. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, J.; Wang, Z.; Jia, M.; Lu, T.; Wang, H.; Yue, W.; Zhang, D.; Li, J.; Wang, L. Association between CNTNAP2 polymorphisms and autism: A family-based study in the chinese han population and a meta-analysis combined with GWAS data of psychiatric genomics consortium. Autism Res. 2019, 12, 553–561. [Google Scholar] [CrossRef]
- Murdoch, J.D.; Gupta, A.R.; Sanders, S.J.; Walker, M.F.; Keaney, J.; Fernandez, T.V.; Murtha, M.T.; Anyanwu, S.; Ober, G.T.; Raubeson, M.J.; et al. No evidence for association of autism with rare heterozygous point mutations in Contactin-Associated Protein-Like 2 (CNTNAP2), or in Other Contactin-Associated Proteins or Contactins. PLoS Genet. 2015, 11, e1004852. [Google Scholar] [CrossRef] [Green Version]
- Oshodi, Y.; Ojewunmi, O.; Oshodi, T.; Ijarogbe, G.; Ogun, O.; Aina, O.; Lesi, F. Oxidative stress markers and genetic polymorphisms of glutathione s-transferase T1, M1, and P1 in a subset of children with autism spectrum disorder in Lagos, Nigeria. Niger. J. Clin. Pract. 2017, 20, 1161. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; DeLaTorre, R.; Taylor, H.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Transl. Psychiatry 2013, 3, e273. [Google Scholar] [CrossRef]
- James, S.J.; Rose, S.; Melnyk, S.; Jernigan, S.; Blossom, S.; Pavliv, O.; Gaylor, D.W. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009, 23, 2374–2383. [Google Scholar] [CrossRef]
- Yui, K.; Kawasaki, Y.; Yamada, H.; Ogawa, S. Oxidative stress and nitric oxide in autism spectrum disorder and other neuropsychiatric disorders. CNS Neurol. Disord. Drug Targets 2016, 15, 587–596. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V.; Brown, W.T.; Cohen, I. Oxidative Stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—The antioxidant proteins. Life Sci. 2004, 75, 2539–2549. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Thorsen, M.; Bilenberg, N.; Thorsen, L.; Michel, T.M. Oxidative stress in adults with autism spectrum disorder: A case control study. J. Autism Dev. Disord. 2021, 52, 275–282. [Google Scholar] [CrossRef]
- Imataka, G.; Yui, K.; Shiko, Y.; Kawasaki, Y.; Sasaki, H.; Shiroki, R.; Yoshihara, S. Urinary and plasma antioxidants in behavioral symptoms of individuals with autism spectrum disorder. Front. Psychiatry 2021, 12, 68445. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Impaired synthesis and antioxidant defense of glutathione in the cerebellum of autistic subjects: Alterations in the activities and protein expression of glutathione-related enzymes. Free Radic. Biol. Med. 2013, 65, 488–496. [Google Scholar] [CrossRef]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meguid, N.A.; Dardir, A.A.; Abdel-Raouf, E.R.; Hashish, A. Evaluation of oxidative stress in autism: Defective antioxidant enzymes and increased lipid peroxidation. Biol. Trace Elem. Res. 2010, 143, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Attia, S.M.; Alshammari, M.A.; Alzahrani, K.S.; Bakheet, S.A. Increased oxidative stress in the cerebellum and peripheral immune cells leads to exaggerated autism-like repetitive behavior due to deficiency of antioxidant response in BTBR T + tf/J mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dawson, V.L.; Dawson, T.M. Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther. 2006, 109, 33–41. [Google Scholar] [CrossRef]
- Förstermann, U.; Schmidt, H.H.H.W.; Pollock, J.S.; Sheng, H.; Mitchell, J.A.; Warner, T.D.; Nakane, M.; Murad, F. Isoforms of nitric oxide synthase characterization and purification from different cell types. Biochem. Pharmacol. 1991, 42, 1849–1857. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Kartawy, M.; Amal, H. The role of nitric oxide in brain disorders: Autism spectrum disorder and other psychiatric, neurological, and neurodegenerative disorders. Redox Biol. 2020, 34, 101567. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M. Chapter 15 nitric oxide in neurodegeneration. Prog. Brain Res. 1998, 118, 215–229. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H.; Kang, G. Nitrosative stress and human disease: Therapeutic potential of denitrosylation. Int. J. Mol. Sci. 2021, 22, 9794. [Google Scholar] [CrossRef]
- Zoroğlu, S.S.; Yürekli, M.; Meram, İ.; Söğüt, S.; Tutkun, H.; Yetkїn, Ö.; Sїvasli, E.; Savaş, H.A.; Yanik, M.; Herken, H.; et al. Pathophysiological role of nitric oxide and adrenomedullin in autism. Cell Biochem. Funct. 2003, 21, 55–60. [Google Scholar] [CrossRef]
- Yao, L.; Fu, H.; Bai, L.; Deng, W.; Xie, F.; Li, Y.; Zhang, R.; Xu, X.; Wang, T.; Lai, S.; et al. Saliva nitrite is higher in male children with autism spectrum disorder and positively correlated with serum nitrate. Redox Rep. 2021, 26, 124–133. [Google Scholar] [CrossRef]
- Wu, S.; Yin, Y.; Du, L. Blood–brain barrier dysfunction in the pathogenesis of major depressive disorder. Cell. Mol. Neurobiol. 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, R.S.; Davies, C.; Iyengar, R.; Perez, L.; Mittal, R.; Eshraghi, A.A. Gut-induced inflammation during development may compromise the blood-brain barrier and predispose to autism spectrum disorder. J. Clin. Med. 2020, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.A.; Iyengar, R.; Eshraghi, R.S.; Bencie, N.; Mittal, J.; Aljohani, A.; Mittal, R.; Eshraghi, A.A. Gene therapy for neurological disorders: Challenges and recent advancements. J. Drug Target 2019, 28, 111–128. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Goines, P.; Van de Water, J. The immune system’s role in the biology of autism. Curr. Opin. Neurol. 2010, 23, 111117. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.-M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Möhrle, D.; Wang, W.; Whitehead, S.N.; Schmid, S. GABAB receptor agonist r-baclofen reverses altered auditory reactivity and filtering in the cntnap2 knock-out rat. Front. Integr. Neurosci. 2021, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, D.; Kabitzke, P.; He, D.; Cox, K.; Thiede, L.; Hanania, T.; Sabath, E.; Alexandrov, V.; Saxe, M.; Peles, E.; et al. Comprehensive analysis of the 16p11.2 deletion and null Cntnap2 mouse models of autism spectrum disorder. PLoS ONE 2015, 10, e0134572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.E.; Kazazian, K.; Mann, R.S.; Möhrle, D.; Schormans, A.L.; Schmid, S.; Allman, B.L. Loss of Cntnap2 in the rat causes autism-related alterations in social interactions, stereotypic behavior, and sensory processing. Autism Res. 2020, 13, 1698–1717. [Google Scholar] [CrossRef]
- Dawes, J.M.; Weir, G.A.; Middleton, S.J.; Patel, R.; Chisholm, K.I.; Pettingill, P.; Peck, L.J.; Sheridan, J.; Shakir, A.; Jacobson, L.; et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron 2018, 97, 806–822.e10. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, R.; Northrop, N.; Yamamoto, B. Fluorescein isothiocyanate (FITC)-dextran extravasation as a measure of blood-brain barrier permeability. Curr. Protoc. Neurosci. 2017, 79, 9–58. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.; Mittal, R.; Shahal, D.; Sinha, P.; Bulut, E.; Mittal, J.; Eshraghi, A.A. Evaluating the Efficacy of taurodeoxycholic acid in providing otoprotection using an in vitro model of electrode insertion trauma. Front. Mol. Neurosci. 2020, 13, 113. [Google Scholar] [CrossRef]
- Manaenko, A.; Chen, H.; Kammer, J.; Zhang, J.H.; Tang, J. Comparison evans blue injection routes: Intravenous versus intraperitoneal, for measurement of blood–brain barrier in a mice hemorrhage model. J. Neurosci. Methods 2011, 195, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Gupta, S.; Sharma, B. Melatonin receptor and KATP channel modulation in experimental vascular dementia. Physiol. Behav. 2015, 142, 66–78. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Burgmans, S.; Hofman, P.A.M.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Blood–brain barrier impairment in dementia: Current and future in vivo assessments. Neurosci. Biobehav. Rev. 2015, 49, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Daneman, R. Formation and maintenance of the BBB. Mech. Dev. 2015, 138, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Carmichael, S.T. Blood−brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr. Opin. Neurol. 2015, 28, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, A.; Marmarou, A.; Hayasaki, K.; Barzo, P.; Fatouros, P.; Corwin, F.; Marmarou, C.; Dunbar, J. The permissive nature of blood brain barrier (BBB) opening in edema formation following traumatic brain injury. In Brain Edema XI; Springer: Berlin/Heidelberg, Germany, 2000; pp. 125–129. [Google Scholar]
- Banks, W.A.; Kovac, A.; Majerova, P.; Bullock, K.M.; Shi, M.; Zhang, J. Tau proteins cross the blood-brain barrier. J. Alzheimers Dis. 2017, 55, 411–419. [Google Scholar] [CrossRef]
- Pluta, R.; Czuczwar, S.J.; Januszewski, S.; Jabłoński, M. The many faces of post-ischemic tau protein in brain neurodegeneration of the Alzheimer’s disease type. Cells 2021, 10, 2213. [Google Scholar] [CrossRef]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood–brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, L.; Du, N.; Yin, X.; Shao, H.; Yang, L. Ramelteon ameliorates LPS-induced hyperpermeability of the blood-brain barrier (BBB) by activating Nrf2. Inflammation 2021, 44, 1750–1761. [Google Scholar] [CrossRef]
- Jin, Z.; Liang, J.; Kolattukudy, P.E. Tetramethylpyrazine preserves the integrity of blood-brain barrier associated with upregulation of MCPIP1 in a murine model of focal ischemic stroke. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, C.; Yang, Y.; Jia, N.; Sun, Q. Protection of vasoactive intestinal peptide on the blood-brain barrier dysfunction induced by focal cerebral ischemia in rats. J. Stroke Cerebrovasc. Dis. 2022, 31, 106160. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Han, Y.-H.; Wang, H.-C.; Lu, C.-T.; Yu, X.-C.; Zhao, Y.-Z. Intradermal Injection of Icariin-HP-β-cyclodextrin improved traumatic brain injury via the trigeminal epineurium-brain dura pathway. J. Drug Target 2022, 30, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, X.; Wang, C.; Wu, X.; Long, B. Hypercholesterolemia aggravates sevoflurane induced cognitive impairment in aged rats by inducing neurological inflammation and apoptosis. J. Biochem. Mol. Toxicol. 2022, e23009. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Xie, L.; Liu, Y.; Liu, T.; Yang, P.; Hu, J.; Peng, Z.; Luo, K.; Du, M.; Chen, C. Astragalus polysaccharide (APS) exerts protective effect against acute ischemic stroke (AIS) through enhancing M2 micoglia polarization by regulating adenosine triphosphate (ATP)/purinergic receptor (P2X7R) axis. Bioengineered 2022, 13, 4468–4480. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Li, F.; Qian, X.; Hong, Z.; Wu, L.; Jiang, Y.; Hu, H. Therapeutic effects of human umbilical cord mesenchymal stem cell on sepsis-associated encephalopathy in mice by regulating PI3K/AKT Pathway. J. Integr. Neurosci. 2022, 21, 38. [Google Scholar] [CrossRef]
- Wang, Z.; Du, X.; Yu, D.; Yang, Y.; Ma, G.; Jia, X.; Cheng, L. Sufentanil alleviates cerebral ischemia-reperfusion injury by inhibiting inflammation and protecting the blood-brain barrier in rats. Eur. J. Histochem. 2022, 66. [Google Scholar] [CrossRef]
- Young, A.M.H.; Campbell, E.; Lynch, S.; Suckling, J.; Powis, S.J. Aberrant NF-KappaB expression in autism spectrum condition: A mechanism for neuroinflammation. Front. Psychiatry 2011, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Kameno, Y.; Iwata, K.; Matsuzaki, H.; Miyachi, T.; Tsuchiya, K.J.; Matsumoto, K.; Iwata, Y.; Suzuki, K.; Nakamura, K.; Maekawa, M.; et al. Serum levels of soluble platelet endothelial cell adhesion molecule-1 and vascular cell adhesion molecule-1 are decreased in subjects with autism spectrum disorder. Mol. Autism 2013, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Onore, C.E.; Nordahl, C.W.; Young, G.S.; Van de Water, J.A.; Rogers, S.J.; Ashwood, P. Levels of soluble platelet endothelial cell adhesion molecule-1 and p-selectin are decreased in children with autism spectrum disorder. Biol. Psychiatry 2012, 72, 1020–1025. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.J.; Hashimoto, K.; Iwata, Y.; Tsujii, M.; Sekine, Y.; Sugihara, G.; Matsuzaki, H.; Suda, S.; Kawai, M.; Nakamura, K.; et al. Decreased serum levels of platelet-endothelial adhesion molecule (PECAM-1) in subjects with high-functioning autism: A negative correlation with head circumference at birth. Biol. Psychiatry 2007, 62, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Sharma, B. Minocycline ameliorates prenatal valproic acid induced autistic behaviour, biochemistry and blood brain barrier impairments in rats. Brain Res. 2016, 1630, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Campbell, M. Tight junction modulation at the blood-brain barrier: Current and future perspectives. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183298. [Google Scholar] [CrossRef]
- Nusrat, A.; Parkos, C.A.; Verkade, P.; Foley, C.S.; Liang, T.W.; Innis-Whitehouse, W.; Eastburn, K.K.; Madara, J.L. Tight junctions are membrane microdomains. J. Cell Sci. 2000, 113, 1771–1781. [Google Scholar] [CrossRef]
- Krafft, P.R.; Caner, B.; Klebe, D.; Rolland, W.B.; Tang, J.; Zhang, J.H. PHA-543613 preserves blood–brain barrier integrity after intracerebral hemorrhage in mice. Stroke 2013, 44, 1743–1747. [Google Scholar] [CrossRef] [Green Version]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative stress in autism spectrum disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative stress and erythrocyte membrane alterations in children with autism: Correlation with clinical features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [Green Version]
- Zoroglu, S.S.; Armutcu, F.; Ozen, S.; Gurel, A.; Sivasli, E.; Yetkin, O.; Meram, I. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur. Arch. Psychiatry Clin. Neurosci. 2004, 254, 143–147. [Google Scholar] [CrossRef]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Aspects Med. 2004, 25, 17–26. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, M.; Hashimoto, T.; Tsuda, Y.; Nakatsu, T.; Kitaoka, T.; Kyotani, S. Assessment of oxidative stress in autism spectrum disorder using reactive oxygen metabolites and biological antioxidant potential. PLoS ONE 2020, 15, e0233550. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder-Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 813304. [Google Scholar] [CrossRef]
- Adams, J.B.; Baral, M.; Geis, E.; Mitchell, J.; Ingram, J.; Hensley, A.; Zappia, I.; Newmark, S.; Gehn, E.; Rubin, R.A.; et al. The severity of autism is associated with toxic metal body burden and red blood cell glutathione levels. J. Toxicol. 2009, 2009, 532640. [Google Scholar] [CrossRef] [Green Version]
- Ghanizadeh, A.; Akhondzadeh, S.; Hormozi, M.; Makarem, A.; Abotorabi-Zarchi, M.; Firoozabadi, A. Glutathione-related factors and oxidative stress in autism, a review. Curr. Med. Chem. 2012, 19, 4000–4005. [Google Scholar] [CrossRef]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (INOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Tsukahara, Y.; Morisaki, T.; Kojima, M.; Uchiyama, A.; Tanaka, M. INOS expression by activated neutrophils from patients with sepsis. ANZ J. Surg. 2001, 71, 15–20. [Google Scholar] [CrossRef]
- Danilov, A.I.; Andersson, M.; Bavand, N.; Wiklund, N.P.; Olsson, T.; Brundin, L. Nitric oxide metabolite determinations reveal continuous inflammation in multiple sclerosis. J. Neuroimmunol. 2003, 136, 112–118. [Google Scholar] [CrossRef]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002, 1, 232–241. [Google Scholar] [CrossRef]
- Amal, H.; Barak, B.; Bhat, V.; Gong, G.; Joughin, B.A.; Wang, X.; Wishnok, J.S.; Feng, G.; Tannenbaum, S.R. Shank3 mutation in a mouse model of autism leads to changes in the s-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry 2018, 25, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Tostes, M.H.F.; Teixeira, H.; Gattaz, W.; Brandão, M.A.; Raposo, N.R. Altered neurotrophin, neuropeptide, cytokines and nitric oxide levels in autism. Pharmacopsychiatry 2012, 45, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Kumar, A.; Ladda, R.; Mainali, G.; Aliu, E. Pitt hopkins-like syndrome 1 with novel CNTNAP2 mutation in siblings. Child Neurol Open. 2021, 8, 2329048X211055330. [Google Scholar] [CrossRef] [PubMed]
- Dilmore, A.H.; McDonald, D.; Nguyen, T.T.; Adams, J.B.; Krajmalnik-Brown, R.; Elijah, E.; Dorrestein, P.C.; Knight, R. The Fecal Microbiome and Metabolome of Pitt Hopkins Syndrome, a Severe Autism Spectrum Disorder. mSystems. 2021, 6, e0100621. [Google Scholar] [CrossRef] [PubMed]
- AlBaazi, S.; Shareef, H. Case report: Pitt-Hopkins like syndrome with CNTNAP2 mutation. Egypt. J. Med. Hum. Genet. 2020, 21, 70. [Google Scholar] [CrossRef]
- Canali, G.; Garcia, M.; Hivert, B.; Pinatel, D.; Goullancourt, A.; Oguievetskaia, K.; Saint-Martin, M.; Girault, J.-A.; Faivre-Sarrailh, C.; Goutebroze, L. Genetic Variants in Autism-Related CNTNAP2 Impair Axonal Growth of Cortical Neurons. Hum. Mol. Genet. 2018, 27, 1941–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Memis, I.; Mittal, R.; Furar, E.; White, I.; Eshraghi, R.S.; Mittal, J.; Eshraghi, A.A. Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model. J. Clin. Med. 2022, 11, 2725. https://doi.org/10.3390/jcm11102725
Memis I, Mittal R, Furar E, White I, Eshraghi RS, Mittal J, Eshraghi AA. Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model. Journal of Clinical Medicine. 2022; 11(10):2725. https://doi.org/10.3390/jcm11102725
Chicago/Turabian StyleMemis, Idil, Rahul Mittal, Emily Furar, Isaiah White, Rebecca S. Eshraghi, Jeenu Mittal, and Adrien A. Eshraghi. 2022. "Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model" Journal of Clinical Medicine 11, no. 10: 2725. https://doi.org/10.3390/jcm11102725
APA StyleMemis, I., Mittal, R., Furar, E., White, I., Eshraghi, R. S., Mittal, J., & Eshraghi, A. A. (2022). Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model. Journal of Clinical Medicine, 11(10), 2725. https://doi.org/10.3390/jcm11102725