
Multiple Arterial Dissections and Connective Tissue Abnormalities

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukherjee, D.; Eagle, K.A. Aortic dissection—An update. Curr. Probl. Cardiol. 2005, 30, 287–325. [Google Scholar] [CrossRef] [PubMed]
- Engelter, S.T.; Traenka, C.; Lyrer, P. Dissection of Cervical and Cerebral Arteries. Curr. Neurol. Neurosci. Rep. 2017, 17, 59. [Google Scholar] [CrossRef]
- Gilhofer, T.S.; Saw, J. Spontaneous coronary artery dissection: A review of complications and management strategies. Expert. Rev. Cardiovasc. Ther. 2019, 17, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Grond-Ginsbach, C.; Pjontek, R.; Aksay, S.S.; Hyhlik-Dürr, A.; Böckler, D.; Gross-Weissmann, M.L. Spontaneous arterial dissection: Phenotype and molecular pathogenesis. Cell Mol. Life Sci. 2010, 67, 1799–1815. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- Grond-Ginsbach, C.; Chen, B.; Pjontek, R.; Wiest, T.; Burwinkel, B.; Tchatchou, S.; Krawczak, M.; Schreiber, S.; Brandt, T.; Kloss, M.; et al. Copy number variation in patients with cervical artery dissection. Eur. J. Hum. Genet. 2012, 20, 1295–1299. [Google Scholar] [CrossRef] [Green Version]
- Ginsbach, P.; Chen, B.; Jiang, X.; Engelter, S.T.; Grond-Ginsbach, C. Copy number studies in noisy datasets. Microarrays 2013, 2, 284–303. [Google Scholar] [CrossRef]
- Shi, Y.; Majewski, J. Fishing CNV: A graphical software package for detecting rare copy number variations in exome—Sequencing data. Bioinformatics 2013, 29, 1461–1462. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequqnce variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- McLysaght, A.; Makino, T.; Grayton, H.M.; Tropeano, M.; Mitchell, K.J.; Vassos, E.; Collier, D.A. Ohnologs are overrepresented in pathogenic copy number mutations. Proc. Natl. Acad. Sci. USA 2014, 111, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA 2010, 107, 9270–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelter, S.T.; Grond-Ginsbach, C.; Metso, T.M.; Metso, A.J.; Kloss, M.; Debette, S.; Leys, D.; Grau, A.; Dallongeville, J.; Bodenant, M.; et al. For the Cervical Artery Dissection and Ischemic Stroke Patients (CADISP) Study Group. Cervical Artery Dissection—Trauma and other potential mechanical trigger events. Neurology 2013, 80, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.; Orberk, E.; Weber, R.; Werner, I.; Busse, O.; Muller, B.; Wigger, F.; Grau, A.; Grond-Ginsbach, C.; Hausser, I. Pathogenesis of cervical artery dissections: Association with connective tissue abnormalities. Neurology 2001, 57, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kloss, M.; Grond-Ginsbach, C.; Ringleb, P.; Hausser, I.; Hacke, W.; Brandt, T. Recurrence of cervical artery dissection: An underestimated risk. Neurology 2018, 90, 1372–1378. [Google Scholar] [CrossRef]
- Grond-Ginsbach, C.; Brandt, T.; Kloss, M.; Aksay, S.S.; Lyrer, P.; Traenka, C.; Erhart, P.; Martin, J.J.; Altintas, A.; Siva, A.; et al. Next generation sequencing analysis of patients with familial cervical artery dissection. Eur. Stroke J. 2017, 2, 137–143. [Google Scholar] [CrossRef]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Abe, K.; Kosho, T.; Yamaguchi, T. Familial aortic dissection of a young adult caused by MYH11 gene mutation. Ann. Thorac. Surg. 2019, 108, 49. [Google Scholar] [CrossRef] [PubMed]
- Grozeva, D.; Kirov, G.; Conrad, D.F.; Barnes, C.P.; Hurles, M.; Owen, M.J.; O’Donovan, M.C.; Craddock, N. Reduced burden of very large and rare CNVs in bipolar affective disorder. Bipolar Disord. 2013, 15, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Guo, D.C.; Prakash, S.K.; McDonald, M.L.; Johnson, R.J.; Wang, M.; Regalado, E.S.; Russel, L.; Cao, J.M.; Kwartler, C.; et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011, 7, e1002118. [Google Scholar] [CrossRef]
- Grond-Ginsbach, C.; Chen, B.; Krawczak, M.; Pjontek, R.; Ginsbach, P.; Jiang, Y.; Abboud, S.; Arnold, M.L.; Bersano, A.; Brandt, T.; et al. Genetic imbalance in patients with cervical artery dissection. Curr. Genomics. 2017, 18, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debette, S.; Goeggel Simonetti, B.; Schilling, S.; Martin, J.J.; Kloss, M.; Sarikaya, H.; Hausser, I.; Engelter, S.; Metso, T.M.; Pezzini, A.; et al. Familial occurrence and heritable connective tissue disorders in cervical artery dissection. Neurology 2014, 83, 2023–2031. [Google Scholar] [CrossRef] [Green Version]
- De Cario, R.; Sticchi, E.; Lucarini, L.; Attanasio, M.; Nistri, S.; Marcucci, R.; Pepe, G.; Giusti, B. Role of TGFBR1 and TGFBR2 genetic variants in Marfan syndrome. J. Vasc. Surg. 2018, 68, 225–233. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewicz, D. Genetics and hereditary factors of aortic disease: Applying what we know, exploring what we don’t. Endovasc. Today 2019, 18, 83–87. [Google Scholar]
- Witsch, J.; Saad, A.; Parikh, S.; Santosh, B.; Murthy, B.S.; Kamel, H.; Navi, B.B.; Segal, A.Z.; Fink, M.E.; Rutrick, S.B.; et al. Association between cervical artery dissection and aortic dissection. Circulation 2021, 144, 840–842. [Google Scholar] [CrossRef]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.; Majesky, M.W.; Daugherty, A. Smooth muscle cells derived from the cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta—Brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; DeRuiter, M.C.; Bergwerff, M.; Poelmann, R.E. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1589–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y. Dorsal aorta formation: Separate origins, lateral-to-medial migration, and remodeling. Develop. Growth Differ. 2013, 55, 113–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvernogeau, L.; Auda-Boucher, G.; Fontaine-Perus, J. Limb bud colonization by somite-derived angioplasty is a crucial step for myoblast emigration. Development 2012, 139, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Que, J.; Wilm, B.; Hasegawa, H.; Wang, F.; Bader, D.M.; Hogan, B.L.M. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc. Natl. Acad. Sci. USA 2008, 105, 16626–16630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilm, B.; Ipenberg, A.; Hastie, N.D.; Burch, J.B.; Bader, D.M. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 2005, 132, 5317–5328. [Google Scholar] [CrossRef] [Green Version]
- Pfaltzgraff, E.R.; Bader, D. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev. Dyn. 2015, 244, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majesky, M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Throm. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.K.; Nocera, A.L.; Dillon, S.T.; Libermann, T.A.; Wendler, O.; Bleier, B.S. Tissue and Exosomal Serine Protease Inhibitors Are Significantly Overexpressed in Chronic Rhinosinusitis with Nasal Polyps. Am. J. Rhinol. Allergy 2019, 33, 359–368. [Google Scholar] [CrossRef]
- Lee, N.H.; Park, S.R.; Lee, J.W.; Lim, S.; Lee, S.H.; Nam, S.; Kim, D.Y.; Hah, S.Y.; Hong, I.S.; Lee, H.Y. SERPINB2 Is a Novel Indicator of Cancer Stem Cell Tumorigenicity in Multiple Cancer Types. Cancers 2019, 11, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzella, J.M.; Frank, M.; Collignon, P.; Langeois, M.; Legrand, A.; Jeunemaitre, X.; Albuisson, J. Phenotypic variability and diffuse arterial lesions in a family with Loeys-Dietz syndrome type 4. Clin. Genet. 2017, 91, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Shalata, A.; Mahroom, M.; Milewicz, D.M.; Limin, G.; Kassum, F.; Badarna, K.; Tarabeih, N.; Assy, N.; Fell, R.; Cohen, H.; et al. Fatal thoracic aortic aneurysm and dissection in a large family with a novel MYLK gene mutation: Delineation of the clinical phenotype. Orphanet. J. Rare Dis. 2018, 13, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riambau, V.; Böckler, D.; Brunkwall, J.; Cao, P.; Chiesa, R.; Coppi, G.; Czerny, M.; Fraedrich, G.; Haulon, S.; Jacobs, M.J.; et al. Editor’s Choice—Management of Descending Thoracic Aorta Diseases: Clinical Practice Guidelines of the European Society for Vascular Surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2017, 53, 4–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]

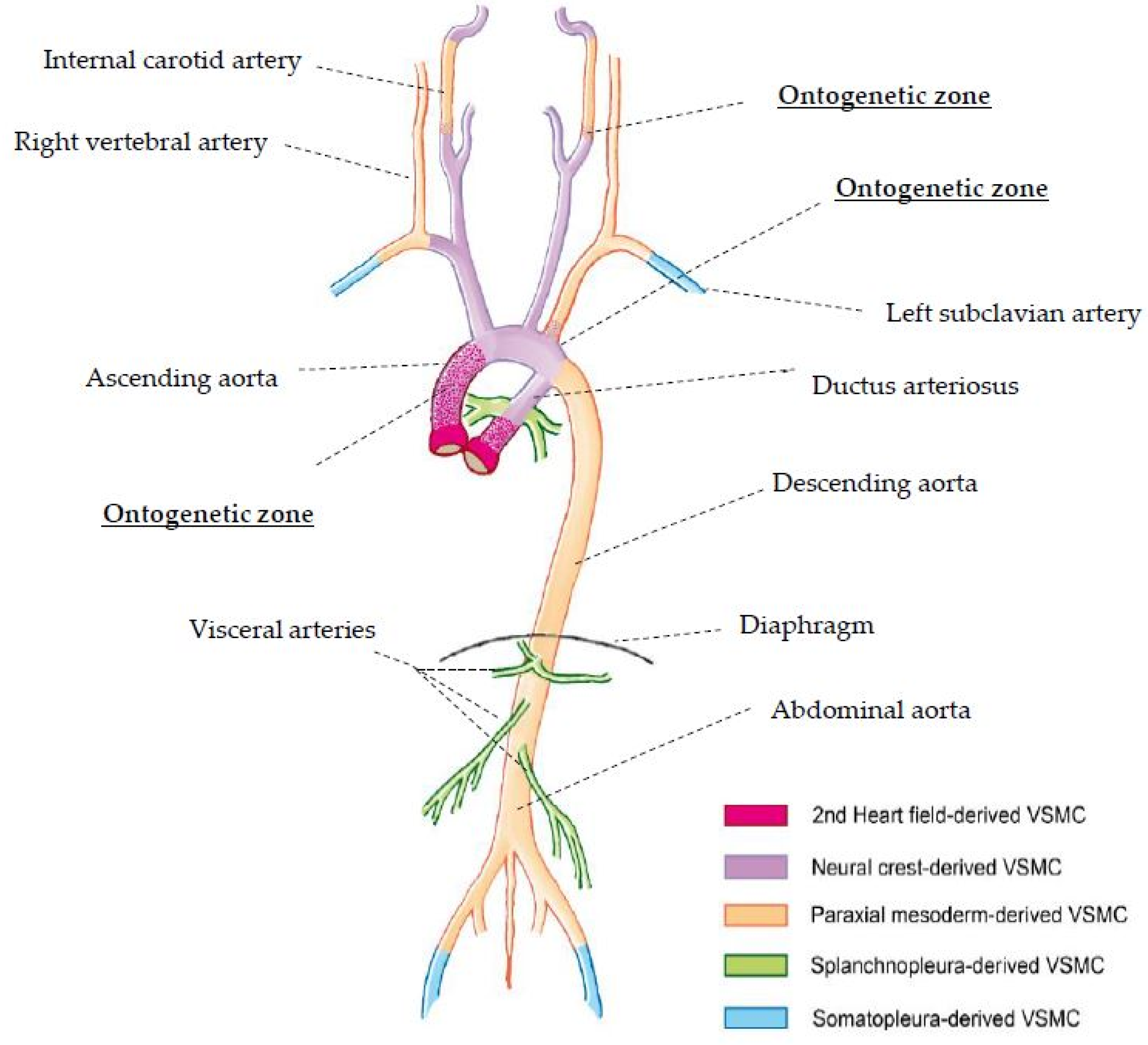

{kind=link}
{kind=link}
| Patient | Sex, Age * | Arterial Locations ** | Phenotype | Familial History |
|---|---|---|---|---|
| 1 | m, 36 | right ICA, aorta type B | normal | positive |
| 2 | m, 39 | bilateral ICA, left VA, aorta type B | normal | positive |
| 3 | m, 45 | left ICA, aorta type A | normal | n.d. |
| 4 | m, 47 | right ICA, aorta type B | hypermobile joints | n.d. |
| Patient | Structural Variant (SV)/Exome Variant (EV) | Nomenclature ISCN2020/HGVS | ACMG Classification | Affected Genes |
|---|---|---|---|---|
| 1 | SV (gain): chr16: 14,916,662–16,306,102 | dup(16)(p13.11) NC_000012.11:g.14916662_16306102dup | Likely pathogenic | MPV17L, C16orf45, MARF1, NDE1, MYH11, FOPNL, ABCC1, ABCC6, NOMO3 |
| 2 | SV (loss): chr2:189.109.859–189.763.802 | del(2)(q32.2)(189401614-190055557) × 1 | Pathogenic | GULP1, DIRC1, COL3A1, COL5A2 |
| 4 | EV: chr2:189.917.732 G/C | gDNA: Chr2(GRCh37):g.189917732G > C | Variant of uncertain significance | COL5A2, amino acid missense substitution Gln856Glu |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erhart, P.; Körfer, D.; Dihlmann, S.; Qiao, J.-L.; Hausser, I.; Ringleb, P.; Männer, J.; Dikow, N.; Schaaf, C.P.; Grond-Ginsbach, C.; et al. Multiple Arterial Dissections and Connective Tissue Abnormalities. J. Clin. Med. 2022, 11, 3264. https://doi.org/10.3390/jcm11123264
Erhart P, Körfer D, Dihlmann S, Qiao J-L, Hausser I, Ringleb P, Männer J, Dikow N, Schaaf CP, Grond-Ginsbach C, et al. Multiple Arterial Dissections and Connective Tissue Abnormalities. Journal of Clinical Medicine. 2022; 11(12):3264. https://doi.org/10.3390/jcm11123264
Chicago/Turabian StyleErhart, Philipp, Daniel Körfer, Susanne Dihlmann, Jia-Lu Qiao, Ingrid Hausser, Peter Ringleb, Jörg Männer, Nicola Dikow, Christian P. Schaaf, Caspar Grond-Ginsbach, and et al. 2022. "Multiple Arterial Dissections and Connective Tissue Abnormalities" Journal of Clinical Medicine 11, no. 12: 3264. https://doi.org/10.3390/jcm11123264
APA StyleErhart, P., Körfer, D., Dihlmann, S., Qiao, J.-L., Hausser, I., Ringleb, P., Männer, J., Dikow, N., Schaaf, C. P., Grond-Ginsbach, C., & Böckler, D. (2022). Multiple Arterial Dissections and Connective Tissue Abnormalities. Journal of Clinical Medicine, 11(12), 3264. https://doi.org/10.3390/jcm11123264