
JAK2 Unmutated Polycythaemia—Real-World Data of 10 Years from a Tertiary Reference Hospital

 , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Cohort Definition
2.2. Laboratory and Clinical Data Source
2.3. Definitions
2.4. Causes of Polycythaemia
2.5. Statistical Analysis
3. Results
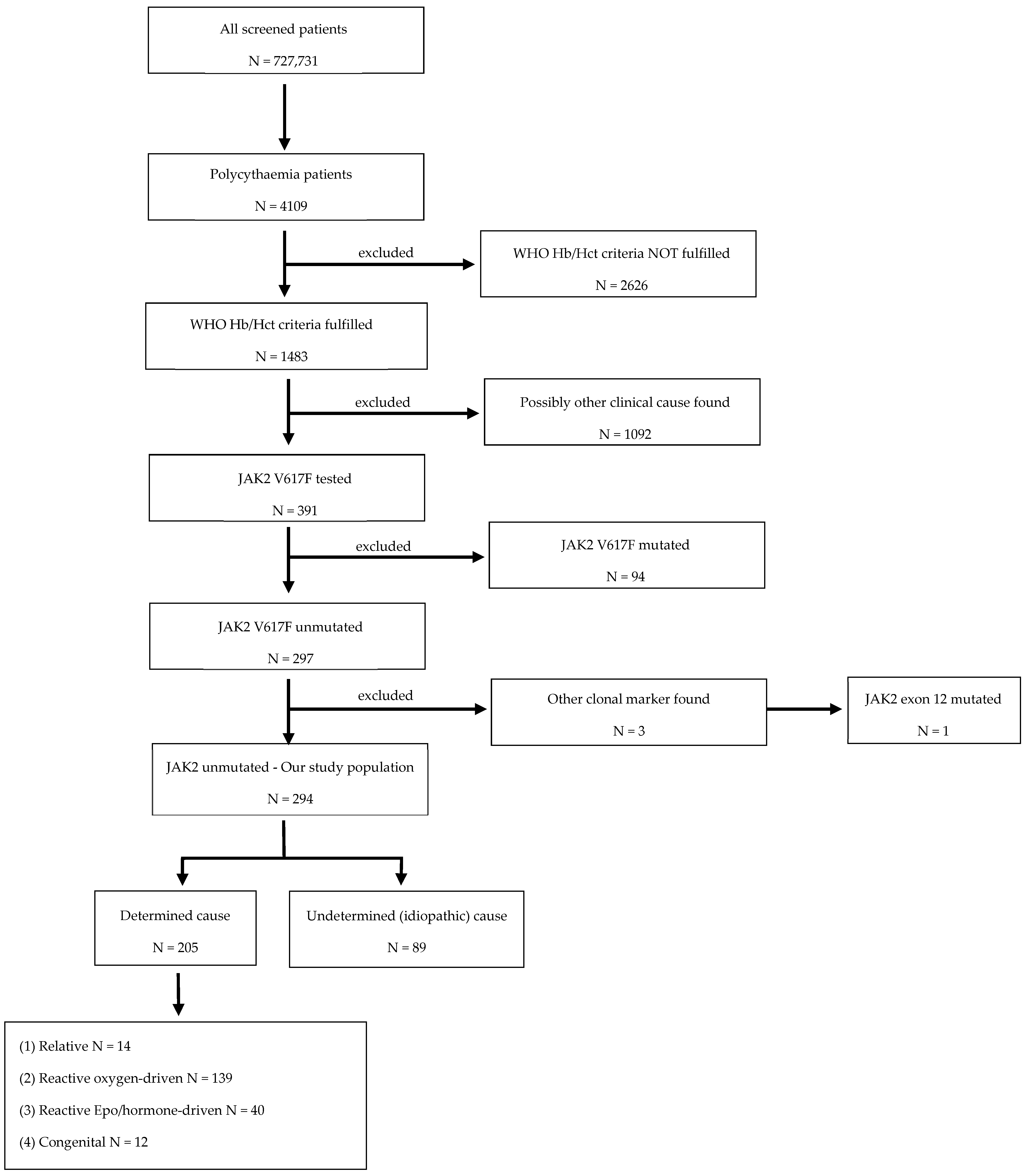
3.1. Study Population
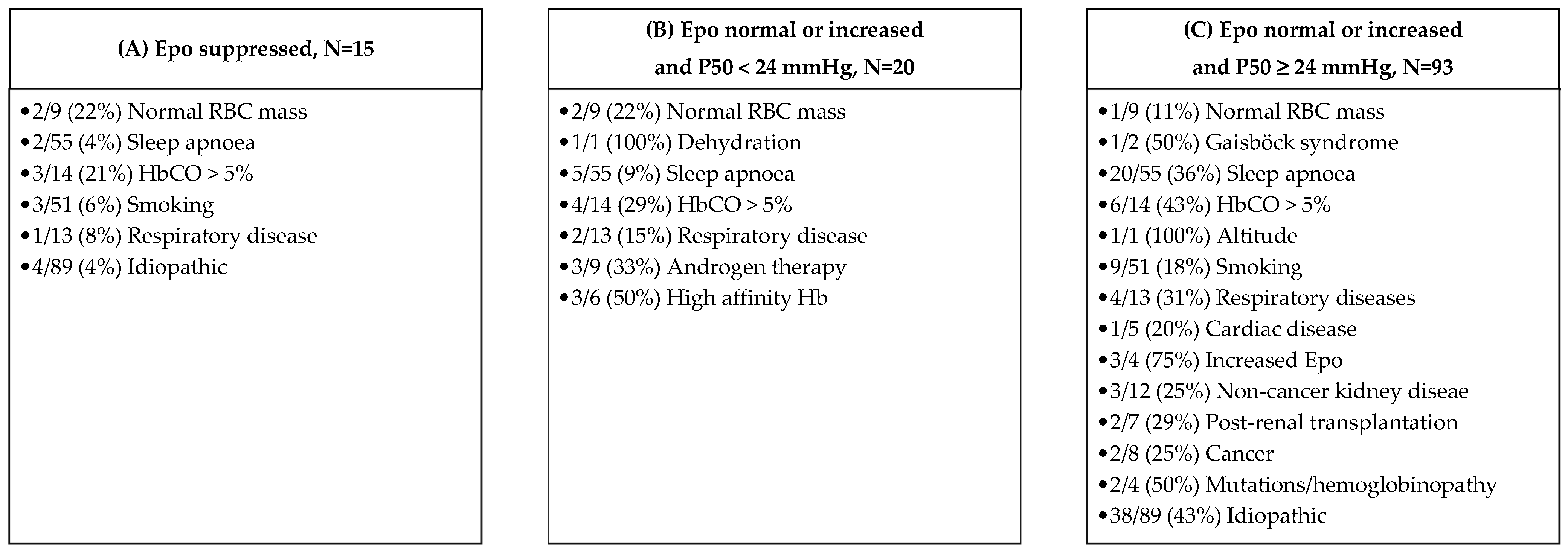
3.2. Causes of JAK2 Unmutated Polycythaemia
3.3. Demographics and Laboratory Values
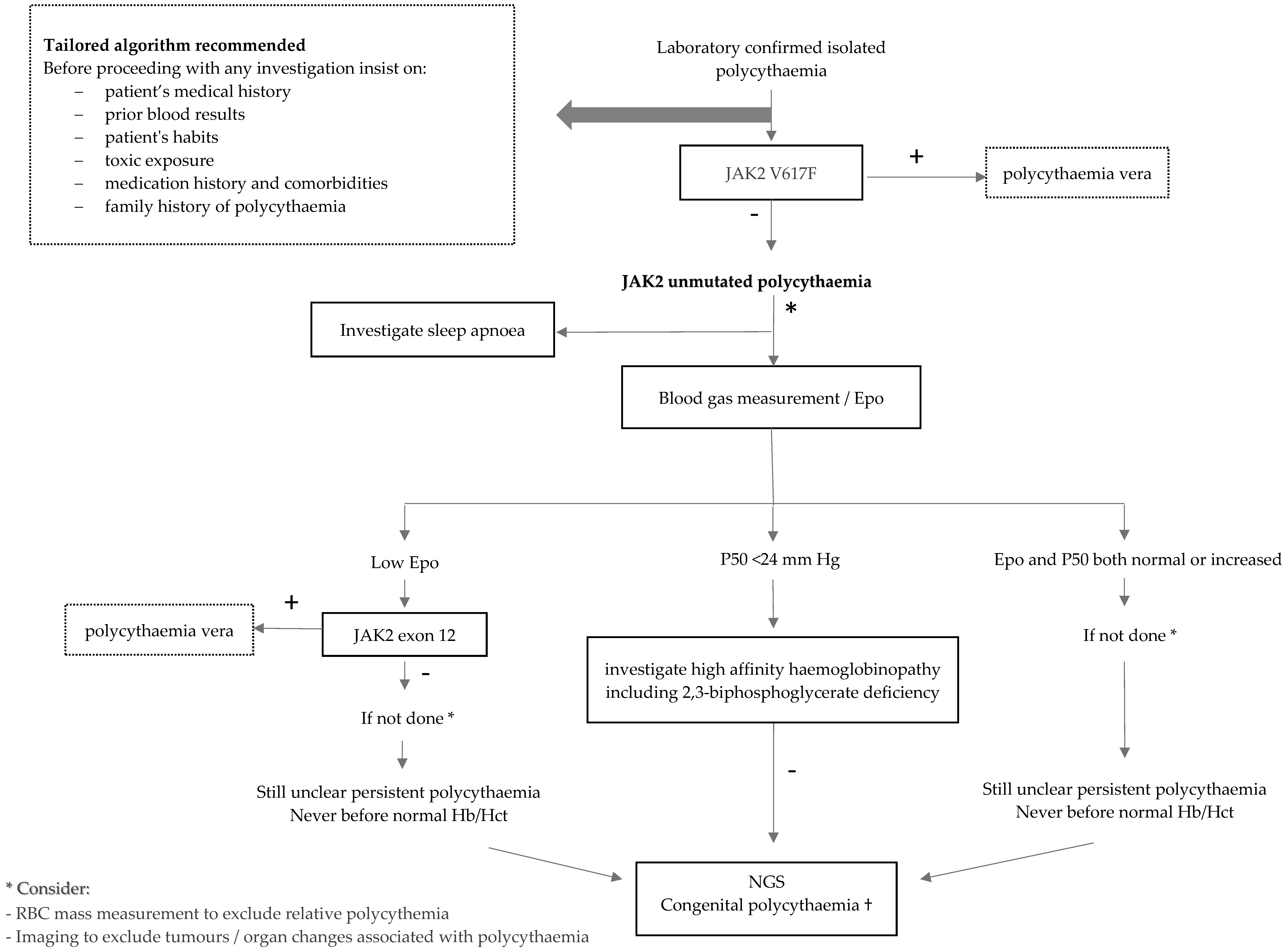
3.4. Investigations of JAK2 Unmuteded Polycythemia
3.5. Management of JAK2 Unmutated Polycythaemia
3.6. Thromboembolic Events in JAK2 Unmutated Patients
3.7. Mortality of JAK2 Unmutated Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Szuber, N.; Pardanani, A.; Tefferi, A. JAK2 unmutated erythrocytosis: Current diagnostic approach and therapeutic views. Leukemia 2021, 35, 2166–2181. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.L.; Coon, L.M.; Frederick, L.A.; Hein, M.; Swanson, K.C.; Savedra, M.E.; Porter, T.R.; Patnaik, M.M.; Tefferi, A.; Pardanani, A.; et al. Genotype–phenotype correlation of hereditary erythrocytosis mutations, a single center experience. Am. J. Hematol. 2018, 93, 1029–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anžej Doma, S.; Drnovšek, E.; Kristan, A.; Fink, M.; Sever, M.; Podgornik, H.; Belčič Mikič, T.; Debeljak, N.; Preložnik Zupan, I. Diagnosis and management of non-clonal erythrocytosis remains challenging: A single centre clinical experience. Ann. Hematol. 2021, 100, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Krečak, I.; Holik, H.; Zekanović, I.; Morić Perić, M.; Marketin, T.; Coha, B.; Gverić-Krečak, V.; Vodanović, M.; Lucijanić, M. Thrombotic risk in secondary polycythemia resembles low-risk polycythemia vera and increases in specific subsets of patients. Thromb. Res. 2022, 209, 47–50. [Google Scholar] [CrossRef]
- Guo, R.; Liang, Y.; Yan, L.; Xu, Z.; Ren, J. Erythrocytosis caused by giant chromophobe renal cell carcinoma: A case report indicating a 9-year misdiagnosis of polycythemia vera. Chin. J. Cancer 2017, 36, 72. [Google Scholar] [CrossRef] [Green Version]
- Kopel, J.; Sharma, P.; Warriach, I.; Swarup, S. Polycythemia with Renal Cell Carcinoma and Normal Erythropoietin Level. Case Rep. Urol. 2019, 2019, 3792514. [Google Scholar] [CrossRef]
- Hama, Y.; Kaji, T.; Ito, K.; Hayakawa, M.; Tobe, M.; Kosuda, S. Erythropoietin-producing renal cell carcinoma arising from autosomal dominant polycystic kidney disease. Br. J. Radiol. 2005, 78, 269–271. [Google Scholar] [CrossRef]
- Adamidou, F.; Mintziori, G.; Vlahaki, E.; Kambaroudis, A. Testosterone and cortisol co-secretion by an adrenocortical adenoma presenting as secondary polycythemia. In Endocrine Abstracts; Bioscientifica: Bristol, UK, 2016; Volume 41. [Google Scholar] [CrossRef] [Green Version]
- Drénou, B.; Le Tulzo, Y.; Caulet-Maugendre, S.; Le Guerrier, A.; Leclercq, C.; Guilhem, I.; Lecoq, N.; Fauchet, R.; Thomas, R. Pheochromocytoma and secondary erythrocytosis: Role of tumour erythropoietin secretion. Nouv. Rev. Française D’hématologie 1995, 37, 197–199. [Google Scholar]
- Sachot, J.L.; Ratajczak, A.; Kerbrat, P.; Leblay, R.; Boffa, G.A. Secondary paraneoplastic polycythemia of testicular cancer. Ann. Urol. 1989, 23, 153–155. [Google Scholar]
- Yoshida, M.; Koshiyama, M.; Fujii, H.; Konishi, M. Erythrocytosis and a fibroid. Lancet 1999, 354, 216. [Google Scholar] [CrossRef]
- Shah, P.C.; Patel, A.R.; Dimaria, F.; Raba, J.; Vohra, R.M. Polycythaemia in lung cancer. Clin. Lab. Haematol. 1979, 1, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Trimble, M.; Caro, J.; Talalla, A.; Brain, M. Secondary erythrocytosis due to a cerebellar hemangioblastoma: Demonstration of erythropoietin mRNA in the tumor. Blood 1991, 78, 599–601. [Google Scholar] [CrossRef] [Green Version]
- Muta, H.; Funakoshi, A.; Baba, T.; Uike, N.; Wakasugi, H.; Kozuru, M.; Jimi, A. Gene expression of erythropoietin in hepatocellular carcinoma. Intern. Med. 1994, 33, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Jalowiec, K.A.; Vrotniakaite-Bajerciene, K.; Capraru, A.; Wojtovicova, T.; Joncourt, R.; Rovó, A.; Porret, N.A. NGS Evaluation of a Bernese Cohort of Unexplained Erythrocytosis Patients. Genes 2021, 12, 1951. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Mehta, J.; Wang, H.; Iqbal, S.U.; Mesa, R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk. Lymphoma 2014, 55, 595–600. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Camps, C.; Petousi, N.; Bento, C.; Cario, H.; Copley, R.R.; McMullin, M.F.; van Wijk, R.; Ratcliffe, P.J.; Robbins, P.A.; Taylor, J.C. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 2016, 101, 1306–1318. [Google Scholar] [CrossRef]
- Bento, C. Genetic basis of congenital erythrocytosis. Int. J. Lab. Hematol. 2018, 40 (Suppl. 1), 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, K.; Colah, R.; Choudhry, V.; Das, R.; Manglani, M.; Madan, N.; Saxena, R.; Jain, D.; Marwaha, N.; Mohanty, D.; et al. Guidelines for screening, diagnosis and management of hemoglobinopathies. Indian J. Hum. Genet. 2014, 20, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, V.; Guitton, C.; Thuret, I.; Rose, C.; Bendelac, L.; Ghazal, K.; Aguilar-Martinez, P.; Badens, C.; Barro, C.; Bénéteau, C.; et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: A retrospective series of 126 patients. Haematologica 2019, 104, 1554–1564. [Google Scholar] [CrossRef] [Green Version]
- Filser, M.; Giansily-Blaizot, M.; Grenier, M.; Monedero Alonso, D.; Bouyer, G.; Pérès, L.; Egée, S.; Aral, B.; Airaud, F.; Da Costa, L.; et al. Increased incidence of germline PIEZO1 mutations in individuals with idiopathic erythrocytosis. Blood 2021, 137, 1828–1832. [Google Scholar] [CrossRef]
- Randi, M.L.; Bertozzi, I.; Cosi, E.; Santarossa, C.; Peroni, E.; Fabris, F. Idiopathic erythrocytosis: A study of a large cohort with a long follow-up. Ann. Hematol. 2016, 95, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Montagnana, M.; Favaloro, E.J.; Franchini, M.; Guidi, G.C.; Lippi, G. The role of ethnicity, age and gender in venous thromboembolism. J. Thromb. Thrombolysis 2010, 29, 489–496. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Polycythaemia | N, All | % Male | Age, Median Years (Range) | Hb, Median g/L (Range) | Hct, Median T/T (Range) | RBC, Median 1012/L (Range) | |
|---|---|---|---|---|---|---|---|
| All | 294 | 82 | 46 (15, 89) | 172 (157, 224) | 0.51 (0.45, 0.68) | 5.67 (4.63, 8.65) | |
| 1. Relative (group 1) | 14 | 71 | 55 (22, 70) | 170 (163, 182) | 0.5 (0.47, 0.65) | 5.67 (4.92, 7.49) | |
| 1.1. Normal RBC mass | 9 | 67 | 59 (25, 68) | 169 (163, 182) | 0.5 (0.47, 0.57) | 5.69 (4.95, 6.69) | |
| 1.2. Dehydration (clinical diagnosis) | 1 | 100 | 22 | 176 | 0.51 | 5.79 | |
| 1.3. Gaisböck syndrome | 2 | 50 | 49 (35, 63) | 175 (170, 180) | 0.52 (0.5, 0.53) | 5.23 (4.92, 5.53) | |
| 1.4. Capillary leak syndrome | 2 | 100 | 54 (37, 70) | 170 (170, 170) | 0.58 (0.5, 0.65) | 6.57 (5.64, 7.49) | |
| 2. Reactive oxygen driven (group 2) | 139 | 80 | 47 (17, 89) | 175 (157, 224) | 0.51 (0.46, 0.68) | 5.71 (4.72, 8.66) | |
| 2.1. Sleep apnoea | 55 | 89 | 49 (20, 89) | 176 (161, 199) | 0.52 (0.47, 0.59) | 5.77 (4.85, 7.06) | |
| 2.2. HbCO >5% | 14 | 64 | 52 (18, 69) | 176 (157, 203) | 0.52 (0.46, 0.58) | 5.94 (4.72, 6.67) | |
| 2.3. Altitude | 1 | 100 | 43 | 179 | 0.5 | 6.06 | |
| 2.4. Smoking | 51 | 76 | 45 (18, 71) | 173 (158, 198) | 0.5 (0.46, 0.58) | 5.56 (4.74, 6.89) | |
| 2.5. Respiratory disease | 13 | 69 | 47 (17, 72) | 173 (165, 207) | 0.51 (0.47, 0.68) | 5.84 (5.21, 8.66) | |
| 2.6. Cardiac disease | 5 | 80 | 40 (21, 50) | 184 (163, 224) | 0.53 (0.49, 0.67) | 5.43 (5.09, 6.6) | |
| 3. Reactive Epo/hormonal driven (group 3) | 40 | 90 | 55 (23, 82) | 171 (164, 199) | 0.51 (0.46, 0.6) | 5.58 (4.86, 6.68) | |
| 3.1. Increased Epo | 4 | 75 | 68 (31, 76) | 174 (166, 199) | 0.53 (0.5, 0.6) | 5.55 (4.86, 6.03) | |
| 3.2. Noncancer kidney disease | 12 | 100 | 52 (23, 76) | 173 (166, 191) | 0.52 (0.48, 0.56) | 5.52 (5.08, 6.28) | |
| 3.3. Post-renal transplantation | 7 | 100 | 59 (32, 69) | 167 (164, 186) | 0.51 (0.47, 0.53) | 5.56 (5.15, 6.3) | |
| 3.4. Androgen therapy | 9 | 100 | 50 (25, 69) | 173 (166, 195) | 0.52 (0.47, 0.58) | 5.7 (5.32, 6.67) | |
| 3.5. Cancer (renal, adrenal, seminoma, lung, brain) | 8 | 62 | 68 (32, 82) | 179 (164, 191) | 0.49 (0.46, 0.59) | 5.62 (5.18, 6.68) | |
| 4. Congenital (group 4) | 12 | 75 | 33 (16, 71) | 172 (161, 184) | 0.51 (0.47, 0.54) | 5.45 (4.63, 7.33) | |
| 4.1. Mutations/hemoglobinopathy | 4 | 100 | 29 (22, 34) | 172 (162, 182) | 0.52 (0.48, 0.54) | 5.90 (5.48, 7.33) | |
| 4.2. Down’s syndrome | 2 | 50 | 37 (16, 57) | 167 (161, 173) | 0.49 (0.47, 0.5) | 5.0 (4.84, 5.07) | |
| 4.3. High affinity Hb (increased P50) | 6 | 67 | 45 (28, 71) | 172 (161, 184) | 0.51 (0.48, 0.53) | 5.28 (4.63, 5.94) | |
| 5. Undetermined (idiopathic) (group 5) | 89 | 85 | 43 (15, 77) | 171 (159, 193) | 0.5 (0.45, 0.56) | 5.65 (4.87, 6.75) | |
| Type of Polycythaemia | N | Age, Median Years (Range) | Thrombotic Events | Therapy Received | Therapy Ongoing | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All, N | Venous, N | Arterial, N | P, N | ASS, N | CRT, N | P, N | ASS, N | CRT, N | ||||
| All | 294 | 46 (15, 89) | 65 | 33 | 32 | 53 | 19 | 2 | 13 | 12 | 1 | |
| 1. Relative | 14 | 55 (22, 70) | 6 | 2 | 4 | 5 | 2 | 0 | 0 | 1 | 0 | |
| 1.1. Normal RBC mass | 9 | 59 (25, 68) | 0 | 0 | 0 | 5 | 2 | 0 | 0 | 1 | 0 | |
| 1.2. Dehydration (clinical diagnosis) | 1 | 22 | 3 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1.3. Gaisböck syndrome | 2 | 49 (35, 63) | 3 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1.4. Capillary leak syndrome | 2 | 54 (37, 70) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 2. Reactive oxygen driven | 139 | 47 (17, 89) | 32 | 18 | 14 | 20 | 7 | 0 | 4 | 3 | 0 | |
| 2.1. Sleep apnoea | 55 | 49 (20, 89) | 15 | 6 | 9 | 11 | 5 | 0 | 2 | 2 | 0 | |
| 2.2. HbCO >5% | 14 | 52 (18, 69) | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | |
| 2.3. Altitude | 1 | 43 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 2.4. Smoking | 51 | 45 (18, 71) | 10 | 6 | 4 | 5 | 2 | 0 | 1 | 1 | 0 | |
| 2.5. Respiratory disease | 13 | 47 (17, 72) | 5 | 5 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | |
| 2.6. Cardiac disease | 5 | 40 (21, 50) | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 3. Reactive Epo/hormonal driven | 40 | 55 (23, 82) | 7 | 5 | 2 | 6 | 1 | 0 | 2 | 1 | 0 | |
| 3.1. Increased Epo | 4 | 68 (31, 76) | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| 3.2. Noncancer kidney disease | 12 | 52 (23, 76) | 1 | 0 | 1 | 2 | 1 | 0 | 2 | 1 | 0 | |
| 3.3. Post-renal transplantation | 7 | 59 (32, 69) | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| 3.4. Androgen therapy | 9 | 50 (25, 69) | 2 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | |
| 3.5. Cancer (renal, adrenal, seminoma, lung, brain) | 8 | 68 (32, 82) | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 4. Congenital | 12 | 33 (16, 71) | 3 | 1 | 2 | 3 | 1 | 1 | 3 | 0 | 0 | |
| 4.1. Mutations/hemoglobinopathy | 4 | 29 (22, 34) | 0 | 0 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | |
| 4.2. Down’s syndrome | 2 | 37 (16, 57) | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 4.3. High affinity Hb (increased P50) | 6 | 45 (28, 71) | 2 | 0 | 2 | 0 | 1 | 1 | 0 | 0 | 0 | |
| 5. Undetermined (idiopathic) | 89 | 43 (15, 77) | 17 | 7 | 10 | 19 | 8 | 1 | 4 | 7 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalowiec, K.A.; Vrotniakaite-Bajerciene, K.; Jalowiec, J.; Frey, N.; Capraru, A.; Wojtovicova, T.; Joncourt, R.; Angelillo-Scherrer, A.; Tichelli, A.; Porret, N.A.; et al. JAK2 Unmutated Polycythaemia—Real-World Data of 10 Years from a Tertiary Reference Hospital. J. Clin. Med. 2022, 11, 3393. https://doi.org/10.3390/jcm11123393
Jalowiec KA, Vrotniakaite-Bajerciene K, Jalowiec J, Frey N, Capraru A, Wojtovicova T, Joncourt R, Angelillo-Scherrer A, Tichelli A, Porret NA, et al. JAK2 Unmutated Polycythaemia—Real-World Data of 10 Years from a Tertiary Reference Hospital. Journal of Clinical Medicine. 2022; 11(12):3393. https://doi.org/10.3390/jcm11123393
Chicago/Turabian StyleJalowiec, Katarzyna Aleksandra, Kristina Vrotniakaite-Bajerciene, Jakub Jalowiec, Noel Frey, Annina Capraru, Tatiana Wojtovicova, Raphael Joncourt, Anne Angelillo-Scherrer, Andre Tichelli, Naomi Azur Porret, and et al. 2022. "JAK2 Unmutated Polycythaemia—Real-World Data of 10 Years from a Tertiary Reference Hospital" Journal of Clinical Medicine 11, no. 12: 3393. https://doi.org/10.3390/jcm11123393