
Non-Invasive Detection of a De Novo Frameshift Variant of STAG2 in a Female Fetus: Escape Genes Influence the Manifestation of X-Linked Diseases in Females

 , ,
, , 
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA (gDNA) Extraction
2.2. SNP-CGH Array
2.3. Cff-DNA Extraction
2.4. Library Construction
2.5. Sequencing
2.6. Estimation of Cff-DNA Fraction
2.7. Investigation for the Presence of a Mosaic in the Parents
2.8. Data Analysis
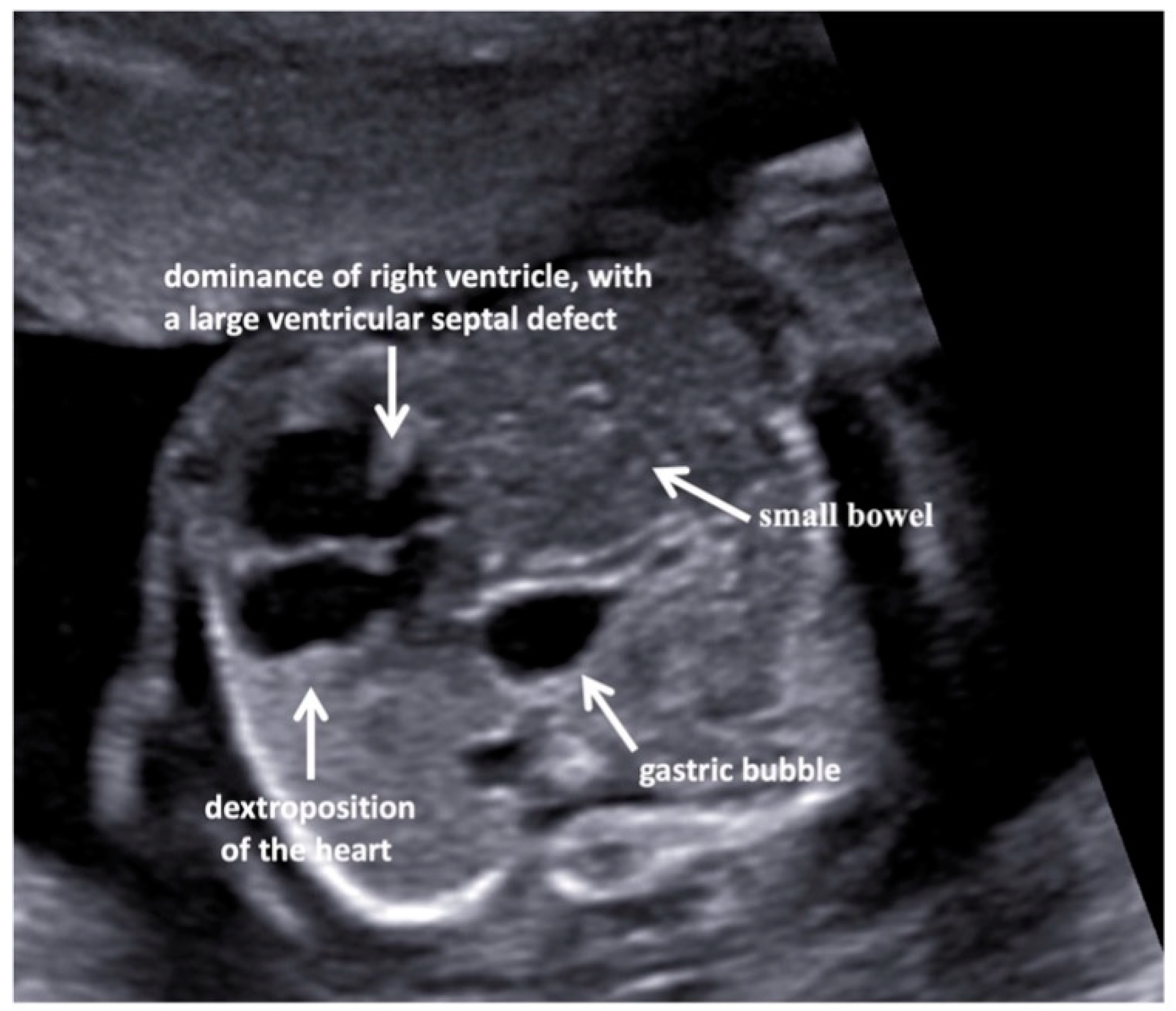
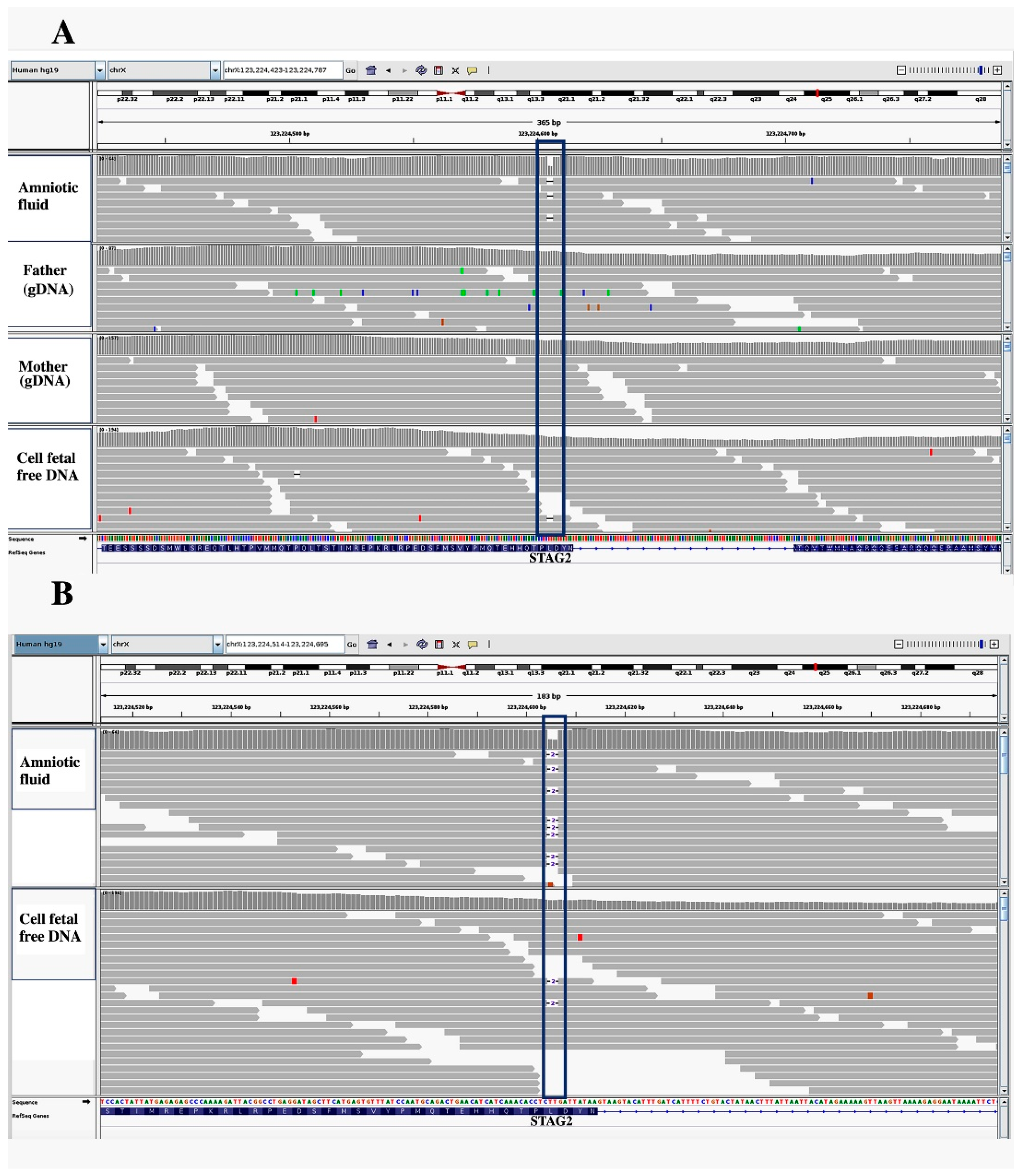
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mone, F.; McMullan, D.J.; Williams, D.; Chitty, L.S.; Maher, E.R.; Kilby, M.D.; Fetal Genomics Steering Group of the British Society for Genetic Medicine; Royal College of Obstetricians and Gynaecologists. Evidence to Support the Clinical Utility of Prenatal Exome Sequencing in Evaluation of the Fetus with Congenital Anomalies: Scientific Impact Paper No. 64 [February] 2021. BJOG 2021, 128, e39–e50. [Google Scholar] [CrossRef]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Scotchman, E.; Shaw, J.; Paternoster, B.; Chandler, N.; Chitty, L.S. Non-invasive prenatal diagnosis and screening for monogenic disorders. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 320–327. [Google Scholar] [CrossRef]
- Provenzano, A.; Palazzo, V.; Reho, P.; Pagliazzi, A.; Marozza, A.; Farina, A.; Zuffardi, O.; Giglio, S. Noninvasive prenatal diagnosis in a family at risk for Fraser syndrome. Prenat. Diagn. 2020, 40, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Saucier, J.B.; Feng, Y.; Jiang, Y.; Sinson, J.; McCombs, A.K.; Schmitt, E.S.; Peacock, S.; Chen, S.; et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat. Med. 2019, 25, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Aoi, H.; Lei, M.; Mizuguchi, T.; Nishioka, N.; Goto, T.; Miyama, S.; Suzuki, T.; Iwama, K.; Uchiyama, Y.; Mitsuhashi, S.; et al. Nonsense variants of STAG2 result in distinct congenital anomalies. Hum. Genome Var. 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Blok, L.S.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.F.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Ragge, N.; Isidor, B.; Bitoun, P.; Odent, S.; Giurgea, I.; Cogné, B.; Deb, W.; Vincent, M.; Jall, J.L.; Morton, J.; et al. Expanding the phenotype of the X-linked BCOR microphthalmia syndromes. Hum. Genet. 2019, 138, 1051–1069. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.O.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef] [Green Version]
- Pezzella, N.; Bove, G.; Tammaro, R.; Franco, B. OFD1: One gene, several disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Garieri, M.; Stamoulis, G.; Blanc, X.; Falconnet, E.; Ribaux, P.; Borel, C.; Santoni, F.; Antonarakis, S.E. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, 13015–13020. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Peng, J.; Baxter, S.; Peng, Z. AutoPVS1: An automatic classification tool for PVS1 interpretation of null variants. Hum. Mutat. 2020, 41, 1488–1498. [Google Scholar] [CrossRef]
- Nishiyama, T. Cohesion and cohesin-dependent chromatin organization. Curr. Opin. Cell Biol. 2019, 58, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Casa, V.; Gines, M.M.; Gusmao, E.G.; Gusmao, E.G.; Slotman, J.A.; Zirkel, A.; Josipovic, N.; Oole, E.; van IJcken, W.F.J.; Houtsmuller, A.B.; et al. Redundant and specific roles of cohesin STAG subunits in chromatin looping and transcriptional control. Genome Res. 2020, 30, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Pérez, L.; Surdez, D.; Brunet, E.; Delattre, O.; Grünewald, T.G.P. STAG Mutations in Cancer. Trends Cancer 2019, 5, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Klein, S.D.; Signer, R.H.; UCLA Clinical Genomics Center; Vilain, E.; Martinez-Agosto, J.A. Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males. Mol. Genet. Genom. Med. 2019, 7, e00501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P.; et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 2019, 21, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hernan, R.R.; Wynn, J.; Chung, W.K. The influence of genetics in congenital diaphragmatic hernia. Semin. Perinatol. 2020, 44, 151169. [Google Scholar] [CrossRef] [PubMed]
- Longoni, M.; High, F.A.; Qi, H.; Joy, M.P.; Hila, R.; Coletti, C.M.; Wynn, J.; Loscertales, M.; Shan, L.; Bult, C.J.; et al. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum. Genet. 2017, 136, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.M.; Campbell, I.M.; Hernandez-Garcia, A.; Lalani, S.R.; Liu, P.; Shaw, C.A.; Rosenfeld, J.A.; Scott, D.A. Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. J. Med. Genet. 2022, 59, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Gofin, Y.; Berry, A.M.; Adams, A.D. Underlying genetic etiologies of congenital diaphragmatic hernia. Prenat Diagn. 2022, 42, 373–386. [Google Scholar] [CrossRef]
- Piché, J.; Vliet, P.P.V.; Pucéat, M.; Andelfinger, G. The expanding phenotypes of cohesinopathies: One ring to rule them all! Cell Cycle 2019, 18, 2828–2848. [Google Scholar] [CrossRef]
- Soardi, F.C.; Machado-Silva, A.; Linhares, N.D.; Zheng, G.; Qu, Q.; Pena, H.B.; Martins, T.M.M.; Vieira, H.G.S.; Pereira, N.B.; Melo-Minardi, R.C.; et al. Familial STAG2 Germline Mutation Defines a New Human Cohesinopathy. NPJ Genom. Med. 2017, 2, 7. [Google Scholar] [CrossRef]
- Kuszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Louie, R.J.; et al. Cohesin complex-associated holoprosencephaly. Brain 2019, 142, 2631–2643. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Wendt, K.S.; Watrin, E.; Azzollini, J.; Braunholz, D.; Buiting, K.; Cereda, A.; Engels, H.; et al. Expanding the clinical spectrum of the ‘HDAC8-phenotype’—Implications for molecular diagnostics, counseling and risk prediction. Clin. Genet. 2016, 89, 564–573. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Jolly, L.A.; Parnell, E.; Gardner, A.E.; Corbett, M.A.; Pérez-Jurado, L.A.; Shaw, M.; Lesca, G.; Keegan, C.; Schneider, M.C.; Griffin, E.; et al. Missense variant contribution to USP9X-female syndrome. NPJ Genom. Med. 2020, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Koninck, M.D.; Lapi, E.; Badía-Careaga, C.; Cossío, I.; Giménez-Llorente, D.; Rodríguez-Corsino, M.; Andrada, E.; Hidalgo, A.; Manzanares, M.; Real, F.X.; et al. Essential Roles of Cohesin STAG2 in Mouse Embryonic Development and Adult Tissue Homeostasis. Cell Rep. 2020, 32, 108014. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Langfelder-Schwind, E.; Farrell, M.H. Challenging the dogma of the healthy heterozygote: Implications for newborn screening policies and practices. Mol. Genet. Metab. 2021, 134, 8–19. [Google Scholar] [CrossRef]
- Wright, C.F.; Prigmore, E.; Rajan, D.; Handsaker, J.; McRae, J.; Kaplanis, J.; Fitzgerald, T.W.; FitzPatrick, D.R.; Firth, H.V.; Hurles, M.E.; et al. Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat. Commun. 2019, 10, 2985. [Google Scholar] [CrossRef]
- Cao, Y.; Tokita, M.J.; Chen, E.S.; Ghosh, R.; Chen, T.; Feng, Y.; Gorman, E.; Gibellini, F.; Ward, P.A.; Braxton, A.; et al. A clinical survey of mosaic single nucleotide variants in disease-causing genes detected by exome sequencing. Genome Med. 2019, 11, 48. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Coverage * | % on Target Reads § | 1× | 5× | 10× | 20× | 30× |
|---|---|---|---|---|---|---|---|
| Mother (gDNA °) | 116X | 85.16% | 99.63 | 99.6 | 99.43 | 98.52 | 96.79 |
| Father(gDNA °) | 118X | 85.87% | 99.82 | 99.65 | 99.46 | 99.05 | 97.60 |
| Amniotic fluid(gDNA °) | 70X | 82.40% | 96.65 | 92.79 | 81.76 | 64.20 | 60 |
| cff-DNA # | 167X | 85.34% | 99.65 | 99.50 | 99.12 | 97.62 | 95.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Provenzano, A.; La Barbera, A.; Lai, F.; Perra, A.; Farina, A.; Cariati, E.; Zuffardi, O.; Giglio, S. Non-Invasive Detection of a De Novo Frameshift Variant of STAG2 in a Female Fetus: Escape Genes Influence the Manifestation of X-Linked Diseases in Females. J. Clin. Med. 2022, 11, 4182. https://doi.org/10.3390/jcm11144182
Provenzano A, La Barbera A, Lai F, Perra A, Farina A, Cariati E, Zuffardi O, Giglio S. Non-Invasive Detection of a De Novo Frameshift Variant of STAG2 in a Female Fetus: Escape Genes Influence the Manifestation of X-Linked Diseases in Females. Journal of Clinical Medicine. 2022; 11(14):4182. https://doi.org/10.3390/jcm11144182
Chicago/Turabian StyleProvenzano, Aldesia, Andrea La Barbera, Francesco Lai, Andrea Perra, Antonio Farina, Ettore Cariati, Orsetta Zuffardi, and Sabrina Giglio. 2022. "Non-Invasive Detection of a De Novo Frameshift Variant of STAG2 in a Female Fetus: Escape Genes Influence the Manifestation of X-Linked Diseases in Females" Journal of Clinical Medicine 11, no. 14: 4182. https://doi.org/10.3390/jcm11144182
APA StyleProvenzano, A., La Barbera, A., Lai, F., Perra, A., Farina, A., Cariati, E., Zuffardi, O., & Giglio, S. (2022). Non-Invasive Detection of a De Novo Frameshift Variant of STAG2 in a Female Fetus: Escape Genes Influence the Manifestation of X-Linked Diseases in Females. Journal of Clinical Medicine, 11(14), 4182. https://doi.org/10.3390/jcm11144182