
Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review


Abstract
1. Introduction
2. Methodology
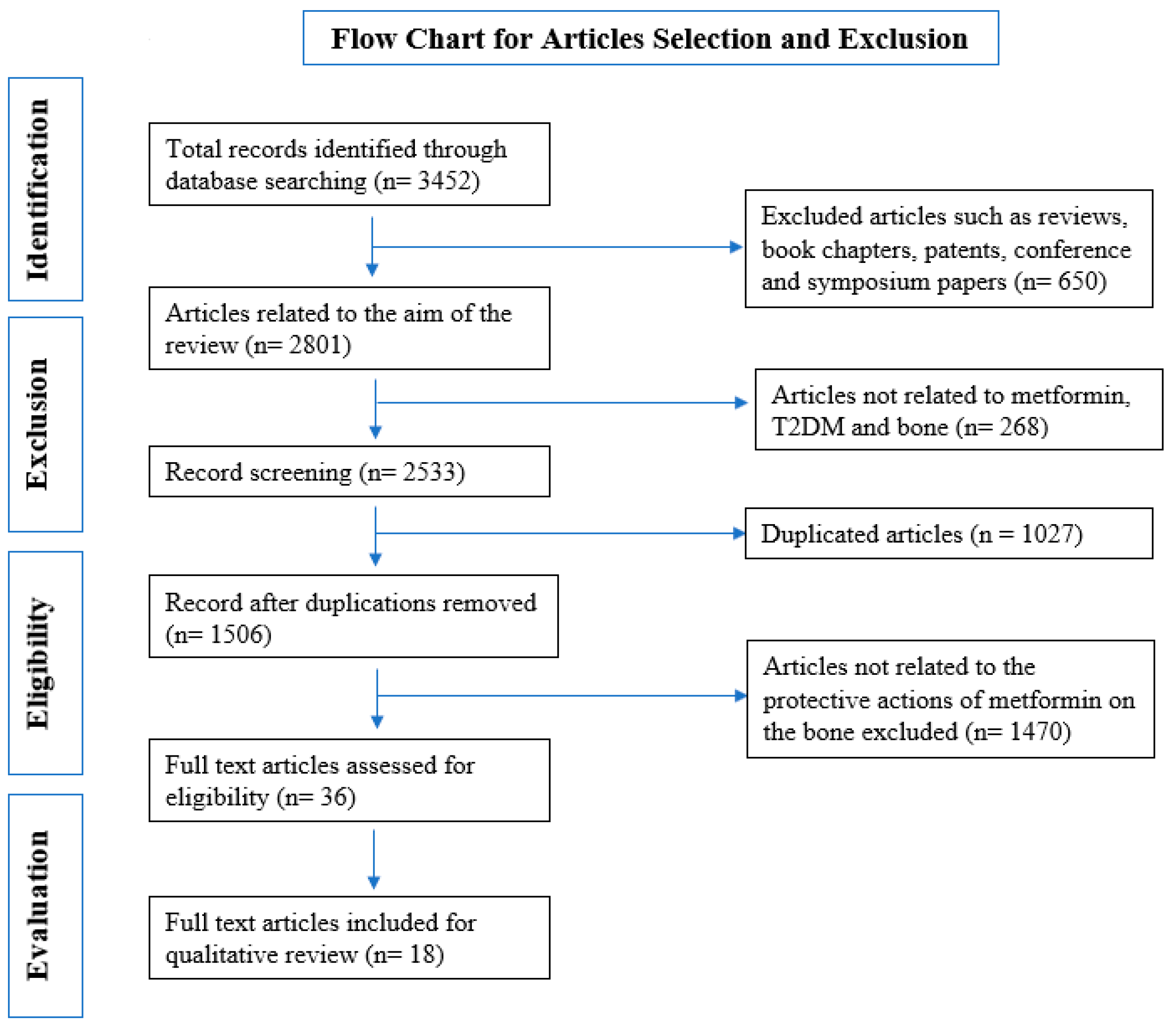
2.1. Selection of Studies
- Yes (the phenotypic feature was present).
- No (the phenotypic feature was not present).
- Not reported (NR; the feature not reported in the model).
- Only significant differences from the control were recorded as demonstrating the phenotype for quantitative data, and the results were then qualitatively assessed.
2.2. Rodent Selection Criteria
3. Results
3.1. Attributes of Subjects
3.2. Nature of the Studies
4. Discussion
4.1. Effects of Type 2 Diabetes Mellitus on Bone Mineral Density
4.2. Effects of Type 2 Diabetes Mellitus on Marrow Adiposity
4.3. Effects of Metformin on Bone Mineral Density
4.4. Effects of Metformin on Marrow Adiposity
5. Strengths and Weaknesses
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Diabetes Federation. Available online: https://idf.org/aboutdiabetes/what-is-diabetes/facts-figures.html (accessed on 4 May 2021).
- National Institutes of Health (NIH). Available online: http://iku.gov.my/nhms-2019 (accessed on 4 May 2021).
- Wang, H.J.; Giambini, H.; Chen, J.W.; Wang, Q.S.; Hou, H.G.; Luo, S.M.; Chen, J.Y.; Zhuang, T.F.; Chen, Y.F.; Wu, T.T.; et al. Diabetes mellitus accelerates the progression of osteoarthritis in streptozotocin-induced diabetic mice by deteriorating bone microarchitecture, bone mineral composition, and bone strength of subchondral bone. Ann. Transl. Med. 2021, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.L.; Ang, S.B.; Chadha, M.; Chow, E.S.L.; Chung, Y.S.; Hew, F.L.; Jaisamrarn, U.; Ng, H.; Takeuchi, Y.; Wu, C.H.; et al. An updated hip fracture projection in Asia: The Asian Federation of Osteoporosis Societies study. Osteoporos. Sarcopenia 2018, 4, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sietsema, D.L. Fighting the Epidemic: Bone Health and Osteoporosis. Nurs. Clin. N. Am. 2020, 55, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr. Diabetes 2019, 20, 5–9. [Google Scholar] [CrossRef]
- Sellmeyer, D.E.; Civitelli, R.; Hofbauer, L.C.; Khosla, S.; Lecka-Czernik, B.; Schwartz, A.V. Skeletal Metabolism, Fracture Risk, and Fracture Outcomes in Type 1 and Type 2 Diabetes. Diabetes 2016, 65, 1757–1766. [Google Scholar] [CrossRef]
- Unnanuntana, A.; Gladnick, B.P.; Donnelly, E.; Lane, J.M. The Assessment of Fracture Risk. J. Bone Jt. Surg. Am. 2010, 92, 743–753. [Google Scholar] [CrossRef]
- Komorita, Y.; Iwase, M.; Fujii, H.; Ohkuma, T.; Ide, H.; Jodai-Kitamura, T.; Sumi, A.; Yoshinari, M.; Nakamura, U.; Kang, D.C.; et al. Serum adiponectin predicts fracture risk in individuals with type 2 diabetes: The Fukuoka Diabetes Registry. Diabetologia 2017, 60, 1922–1930. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamauchi, M.; Sugimoto, T. Prevalent vertebral fracture is dominantly associated with spinal microstructural deterioration rather than bone mineral density in patients with type 2 diabetes mellitus. PLoS ONE 2019, 14, e0222571. [Google Scholar] [CrossRef]
- Yu, E.W.; Greenblatt, L.; Eajazi, A.; Torriani, M.; Bredella, M.A. Marrow adipose tissue composition in adult with morbid obesity. Bone 2017, 97, 38–42. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef]
- American Diabetes Association. Pharmacologic Approaches to Glycemic Treatment. In Standards of Medical Care in Diabetes. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Ji, B.Y.; Zhang, Y.C. Role of Metformin on Osteoblast Differentiation in Type 2 Diabetes. BioMed Res. Int. 2019, 2019, 9203934. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Liello, R.D.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef] [PubMed]
- Bahrambeigi, S.; Yousefi, B.; Rahimi, M.; Shafiei-Irannejad, V. Metformin; an old antidiabetic drug with new potentials in bone disorders. Biomed. Pharmacother. 2019, 109, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Mondockova, V.; Adamkovicova, M.; Lukacova, M.; Grosskopf, B.; Babosova, R.; Galbavy, D.; Martiniakova, M.; Omelka, R. The estrogen receptor 1 gene affects bone mineral density and osteoporosis treatment efficiency in Slovak postmenopausal women. BMC Med. Genet. 2018, 19, 174. [Google Scholar] [CrossRef]
- Finkelstein, J.S.; Brockwell, S.E.; Mehta, V.; Greendale, G.A.; Sowers, M.F.R.; Ettinger, B.; Lo, J.C.; Johnston, J.M.; Cauley, J.A.; Danielson, M.E.; et al. Bone Mineral Density Changes during the Menopause Transition in a Multiethnic Cohort of Women. J. Clin. Endocrinol. Metab. 2008, 93, 861–868. [Google Scholar] [CrossRef]
- Zhu, J.; Ji, M.X.; Xing, L.; Yu, Z.Z.; Guo, X.Y.; Chen, X.P.; Shu, J. Ovarian Hormonal Change-Related Energy Metabolism and Obesity in Menopausal Women. In Hormone Therapy and Replacement in Cancer and Aging-Related Disease; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Adeyemi, W.J.; Olayaki, L.A.; Abdussalam, T.A.; Fabiyi, T.O.; Raji, T.L.; Adetunji, A.A.R. Co-administration of omega-3 fatty acids and metformin showed more desirable effects than the single therapy on indices of bone mineralization but not gluco-regulatory and antioxidant markers in diabetic rats. Biomed. Pharmacother. 2020, 121, 109631. [Google Scholar] [CrossRef]
- Adulyaritthikul, P.; Sanit, J.; Nokkaew, N.; Kongpol, K.; Mongkolpathumrat, P.; Lampang, S.N.; Rojviriya, C.; Kumphune, S. The effect of metformin and P38 MAPK inhibitor on diabetic bone porosity in non-obese type 2 diabetic rats. J. Appl. Pharm. Sci. 2019, 9, 82–90. [Google Scholar] [CrossRef]
- Aljalaud, N.A. Ameliorative and Preventive Effects of Metformin, Nigella sativa, Punica granatum and Zingiber officinale on Bone Damage and Infections Caused by Diabetes mellitus in Animal Model. J. Pure Appl. Microbiol. 2019, 13, 465–473. [Google Scholar] [CrossRef]
- Felice, J.I.; Schurmana, L.; McCarthya, A.M.; Sedlinskya, C.; Aguirreb, J.I.; Cortizo, A.M. Effects of fructose-induced metabolic syndrome on rat skeletal cells and tissue, and their responses to metformin treatment. Diabetes Res. Clin. Pract. 2017, 126, 202–213. [Google Scholar] [CrossRef]
- Bornstein, S.; Moschetta, M.; Kawano, Y.; Sacco, A.; Huynh, D.; Brooks, D.; Manier, S.; Fairfield, H.; Falank, C.; Roccaro, A.M.; et al. Metformin Affects Cortical Bone Mass and Marrow Adiposity in Diet-Induced Obesity in Male Mice. Endocrinology 2017, 158, 3369–3385. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, M.S.; Schurman, L.; McCarthy, A.D.; Cortizo, A.M.; Tolosa, M.J.; Gangoiti, M.V.; Arnol, V.; Sedlinsky, C. Effect of Metformin on Bone Marrow Progenitor Cell Differentiation: In Vivo and In Vitro Studies. J. Bone Miner. Res. 2010, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.d.S.B.F.; Brito, G.A.d.C.; Lima, M.L.dS.; Silva Júnior, A.A.d.; Silva, E.d.S.; de Rezende, A.A.; Bortolin, R.H.; Galvan, M.; Pirih, F.Q.; Araújo Júnior, R.F.d.; et al. Metformin Hydrochloride-Loaded PLGA Nanoparticle in Periodontal Disease Experimental Model Using Diabetic Rats. Int. J. Mol. Sci. 2018, 19, 3488. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Du, J.; Feng, W.; Lu, B.Y.; Liu, H.R.; Guo, J.; Amizuka, N.; Li, M.Q. Histological evidence that metformin reverses the adverse effects of diabetes on orthodontic tooth movement in rats. J. Mol. Hist. 2017, 48, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, M.J.; Chuguransky, S.R.; Sedlinsky, C.; Schurman, L.; McCarthy, A.D.; Molinuevo, M.S.; Cortizo, A.M. Insulin-deficient diabetes-induced bone microarchitecture alterations are associated with a decrease in the osteogenic potential of bone marrow progenitor cells: Preventive effects of metformin. Diabetes Res. Clin. Pract. 2013, 101, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Wang, Q.; Nie, L.L.X.; Zhang, P.; Zhao, P.F.; Yuan, Q.; Ji, N.; Ding, Y.; Wang, Q. Metformin ameliorates the NLPP3 inflammasome mediated pyroptosis by inhibiting the expression of NEK7 in diabetic periodontitis. Arch. Oral Biol. 2020, 116, 104763. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Zhang, P.; Wang, Q.; Ji, N.; Xia, S.S.; Ding, Y.; Wang, Q. Metformin ameliorates experimental diabetic periodontitis independently of mammalian target of rapamycin (mTOR) inhibition by reducing NIMA-related kinase 7 (Nek7) expression. J. Periodontol. 2019, 90, 1032–1042. [Google Scholar] [CrossRef]
- Zheng, L.F.; Shen, X.M.; Ye, J.J.; Xie, Y.; Yan, S.J. Metformin alleviates hyperglycemia-induced apoptosis and differentiation suppression in osteoblasts through inhibiting the TLR4 signaling pathway. Life Sci. 2019, 216, 29–38. [Google Scholar] [CrossRef]
- Lee, H.J.; Cantú, S.M.; Donoso, A.S.; Choi, M.R.; Peredo, H.A.; Puyó, A.M. Metformin prevents vascular prostanoid release alterations induced by a high-fat diet in rats. Auton. Autacoid Pharmacol. 2017, 37, 37–43. [Google Scholar] [CrossRef]
- Kim, E.K.; Lee, S.H.; Jhun, J.Y.; Byun, J.K.; Jeong, J.H.; Lee, S.Y.; Kim, J.K.; Choi, J.Y.; Cho, M.L. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing FGF21. Mediat. Inflamm. 2016, 2016, 5813030. [Google Scholar] [CrossRef]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.L.; Jiang, B.B.; Xu, X.J.; Han, J.Y.; Yan, Y.; Yang, Q.; et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Santana, K.N.; Lelis, D.F.; Mendes, K.L.; Lula, J.F.; Paraíso, A.F.; Andrade, J.F.O.; Feltenberger, J.D.; Cota, J.; da Costa, D.V.; de Paula, A.M.B.; et al. Metformin Reduces Lipogenesis Markers in Obese Mice Fed a Low-Carbohydrate and High-Fat Diet. Lipids 2016, 51, 1375–1384. [Google Scholar] [CrossRef]
- Ismail, T.A.; Soliman, M.M.; Ismail, S.A. Adiponectin Regulation in Type 2 Diabetic Rats: Effects of Insulin, Metformin and dexamethasone. Am. J. Pharmacol. Toxicol. 2013, 8, 197–208. [Google Scholar] [CrossRef]
- Pei, L.; Yang, J.; Du, J.; Liu, H.Q.; Ao, N.; Zhang, Y.Y. Downregulation of chemerin and alleviation of endoplasmic reticulum stress by metformin in adipose tissue of rats. Diabetes Res. Clin. Pract. 2012, 97, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Camacho, P.M.; Petak, S.M.; Binkley, N.; Diab, D.L.; Eldeiry, L.S.; Farooki, A.; Harris, S.T.; Hurley, D.L.; Kelly, J.; Lewiecki, E.M.; et al. American Association of Clinical Endocrinologist/American College of Endocrinology Clinical Practice Guidelines for the Diagnosis and Treatment of Postmenopausal Osteoporosis—2020 Update Executive Summary. Endocr. Prac. 2020, 26, 564–570. [Google Scholar] [CrossRef]
- Trammell, L.H.; Kroman, A.M. Chapter 13-Bone and Dental Histology. In Research Methods in Human Skeletal Biology, 1st ed.; DiGangi, E.A., Moore, M.K., Eds.; Elsevier: Oxford, UK, 2013; pp. 361–395. [Google Scholar]
- Cortet, B.; Lucas, S.; Legroux-Gerot, I.; Penel, G.; Chauveau, C.; Paccou, J. Bone disorders associated with diabetes mellitus and its treatments. Jt. Bone Spine 2019, 86, 315–320. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Picke, A.K.; Campbell, G.; Napoli, N.; Hofbauer, L.C.; Rauner, M. Update on the impact of type 2 diabetes mellitus on bone metabolism and material properties. Endocr. Connect. 2019, 8, R55–R70. [Google Scholar] [CrossRef]
- Hayashi, M.; Ono, T.; Nakashima, T. Signaling in Osteoblast Differentiation. In Physiology and Pathobiology of Bone Formation, 1st ed.; Cao, X., Ed.; Elsevier: Tokyo, Japan, 2020; Volume 2020, pp. 416–426. [Google Scholar] [CrossRef]
- Palomer, X.; Delgado-Pizzaro, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Wang, P.; He, Q.J.; Ma, R.; Lee, S.C. PPARγ promotes diabetes-associated centrosome amplification via increasing the expression of SKA1 directly at the transcriptional level. J. Cell Physiol. 2019, 234, 20694–20703. [Google Scholar] [CrossRef]
- Wang, K.; Sun, J.; Teng, J.F.; Yu, Y.F.; Zhong, D.C.; Fan, Y. Overexpression of spindle and kinetochore–associated protein 1 contributes to the progression of prostate cancer. Tumor Biol. 2017, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Muruganandan, S.; Govindarajan, R.; Sinal, C.J. Bone Marrow Adipose Tissue and Skeletal Health. Curr. Osteoporos. Rep. 2018, 16, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Meng, Y.; Yu, X.J. The Unique Metabolic Characteristics of Bone Marrow Adipose Tissue. Front. Endocrinol. 2019, 10, 69. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications-A Review. Nutr. J. 2014, 13, 1–9. [Google Scholar] [CrossRef]
- Salvatori, R. Growth hormone deficiency in patients with obesity. Endocrine 2015, 49, 304–306. [Google Scholar] [CrossRef]
- Yang, A.; Cho, S.Y.; Kwak, M.J.; Kim, S.J.; Park, S.W.; Jin, D.K.; Lee, J.E. Impact of BMI on peak growth hormone responses to provocative tests and therapeutic outcome in children with growth hormone deficiency. Sci. Rep. 2019, 9, 16181. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A.; Klibanski, A. Chapter Nine-Effects of Growth Hormone on Bone. In Progress in Molecular Biology and Translational Science: Growth Hormone in Health and Disease, 1st ed.; Casanueva, F.F., Ed.; Elsevier: London, UK, 2016; pp. 193–211. [Google Scholar] [CrossRef]
- Agha, A.; Monson, J.P. Modulation of glucocorticoid metabolism by the growth hormone–IGF-1 axis. Clin. Endocrinol. 2007, 66, 459–465. [Google Scholar] [CrossRef]
- Laycock, J.; Meeran, K. Chapter 10: The Adrenal Glands (1): Adrenal Cortex. In Integrated Endocrinology, 1st ed.; John Wiley & Sons: Oxford, UK, 2013; pp. 211–246. [Google Scholar]
- Ahmad, A.M.; Thomas, J.; Clewes, A.; Hopkins, M.T.; Guzder, R.; Ibrahim, H.; Durham, B.H.; Vora, J.P.; Fraser, W.D. Effects of Growth Hormone Replacement on Parathyroid Hormone Sensitivity and Bone Mineral Metabolism. J. Clin. Endocrinol. Metab. 2003, 88, 2860–2868. [Google Scholar] [CrossRef][Green Version]
- Mrak, E.; Villa, I.; Lanzi, R.; Losa, M.; Guidobono, F.; Rubinacci, A. Growth hormone stimulates osteoprotegerin expression and secretion in human osteoblast-like cells. J. Endocrinol. 2007, 192, 639–645. [Google Scholar] [CrossRef]
- Kim, T.Y.; Schafer, A.L. Diabetes and Bone Marrow Adiposity. Curr. Osteoporos. Rep. 2016, 14, 337–344. [Google Scholar] [CrossRef]
- Griffith, M.L.; Younk, L.M.; Davis, S.N. Visceral Adiposity, Insulin Resistance, and Type 2 Diabetes. Am. J. Lifestyle Med. 2010, 4, 230–243. [Google Scholar] [CrossRef]
- Hivert, M.F.; Sullivan, L.M.; Shrader, P.; Fox, C.S.; Nathan, D.M.; D’Agostino, R.B.; Wilson, P.W.F.; Benjamin, E.J.; Meigs, J.B. The association of tumor necrosis factor α receptor 2 and tumor necrosis factor α with insulin resistance and the influence of adipose tissue biomarkers in humans. Metab. Clin. Exp. 2010, 59, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Franco, S.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-α. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279. [Google Scholar] [CrossRef]
- Cersosimo, E.; DeFronzo, R.A. Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes Metab. Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Al-Shoumer, K.A.S.; Al-Essa, T.M. Is there a relationship between vitamin D with insulin resistance and diabetes mellitus? World J. Diabetes 2015, 6, 1057–1064. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.S.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Invest. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schiling, A.F.; Beil, F.T.; Shen, J.H.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin Inhibits Bone Formation through a Hypothalamic Relay: A Central Control of Bone Mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef]
- Roden, M.; Ludwig, C.; Nowotny, P.; Schneider, B.; Clodi, M.; Vierhapper, H.; Roden, A.; Waldhäusl, W. Relative hypoleptinemia in patients with type 1 and type 2 diabetes mellitus. Int. J. Obes. 2000, 24, 976–981. [Google Scholar] [CrossRef][Green Version]
- Pan, H.T.; Guo, J.; Su, Z.Q. Advances in understanding the interrelations between leptin resistance and obesity. Physiol. Behav. 2014, 130, 157–169. [Google Scholar] [CrossRef]
- Hamrick, M.W.; Ferrari, S.L. Leptin and the sympathetic connection of fat to bone. Osteoporos. Int. 2007, 19, 905–912. [Google Scholar] [CrossRef]
- Aung, M.; Amin, S.; Gulraiz, A.; Gandhi, F.R.; Pena Escobar, J.A.; Malik, B.H. The Future of Metformin in the Prevention of Diabetes-Related Osteoporosis. Cureus 2020, 12, e1041. [Google Scholar] [CrossRef] [PubMed]
- İşeri, H.; Kurt, G.; Kişnişci, R. Chapter 25-Biomechanics of Rapid Tooth Movement by Dentoalveolar Distraction Osteogenesis. In Current Therapy in Orthodontics, 1st ed.; Nanda, R., Kapila, S., Eds.; Elsevier: Saint Louis, MO, USA, 2010; pp. 321–337. [Google Scholar] [CrossRef]
- Lin, F.; Pan, Y.N.; Zhang, Y.W.; Zhou, Q. The Effect of Metformin on Vertebral Marrow Fat in Postmenopausal Women with Newly Diagnosed Type 2 Diabetes Mellitus. Menopause 2020, 27, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.L.; Johnson, J.; Vemulapalli, T.H.; Crisler, J.R.; Shepherd, R. Chapter 7–Commonly Used Animal Models. In Principles of Animal Research for Graduate and Undergraduate Students, 1st ed.; Suckow, M.A., Stewart, K.L., Eds.; Academic Press: Boston, MS, USA, 2013; pp. 117–175. [Google Scholar] [CrossRef]
- Kottaisamy, C.P.D.; Raj, D.S.; Kumar, V.P.; Sankaran, U. Experimental animal models for diabetes and its related complications—a review. Lab. Anim. Res. 2021, 37, 23. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Determinants of Bone Mineral Density | Determinants of Adiposity |
|---|---|
| Presence or progression of bone porosity | Decreased adipocyte size |
| Low bone mineral density/mineralization | Decreased fat mass |
| Alkaline phosphatase (ALP) | Total serum triglyceride (TG) |
| Tartrate-resistant acid phosphatase (TRAP) | Total cholesterol (TC) |
| First Author, Year | Mouse Strain | Age | Duration of Treatment | Dosage of Treatment | Determinants of Bone Health | |||
|---|---|---|---|---|---|---|---|---|
| Reduced Bone Porosity | Improved Bone Mineral Density | Increased ALP | Decreased TRAP | |||||
| Adeyemi, 2020 [20] | Wistar | 10-12 weeks old | 4 weeks | 180 and 200 mg/kg/body weight (b.w.)/per day (p.o.) | NR | NR | Yes | Yes |
| Adulyaritthikul, 2019 [21] | Goto-Kakizaki (GK) rats and Wistar (control) | NR | 4 weeks | 15 mg/kg b.w. twice daily (b.i.d) | Yes (cortical, trabecular and iliac bone) | NR | Yes | NR |
| Aljalaud, 2019 [22] | Albino Wistar | 8 weeks old | 6 weeks | 150 mg/kg/day | NR | Yes | NR | NR |
| Felice, 2017 [23] | Wistar | 8 weeks old | 3 weeks | 100 mg/kg/day | NR | Yes | Yes | Yes |
| Bornstein, 2017 [24] | C57BL/6J | 18 weeks old | 6 weeks | 300 mg/kg/day | Yes | Yes | NR | NR |
| Molinuevo, 2010 [25] | Sprague-Dawley | NR | 2 weeks | 100 mg/kg/day | NR | NR | Yes | Yes |
| Pereira, 2018 [26] | Wistar | NR | 10 days | 50 mg/kg/day and 100 mg/kg/day Poly (lac-tic-co-glycolic acid) (PLGA) + 10 mg/kg & 100 mg/kg | Yes | NR | NR | NR |
| Sun, 2017 [27] | Wistar | 7 weeks old | 1 month | 100 mg/kg/bw/day | NR | NR | Yes | Yes |
| Tolosa, 2013 [28] | Sprague-Dawley | 2 months old | 2 weeks | 100 mg/kg/day | NR | NR | Yes | No (Increase) |
| Zhou, 2020 [29] | BKS-Leprem2Cd479 and C57BLKS (control) | 6 weeks old | 9 weeks | 200 mg/kg/day | No (No Changes) | No (No Changes) | NR | NR |
| Zhou, 2019 [30] | C57BL/6 wild-type | 6 weeks old | 10 weeks | 200 mg/kg/day | NR | Yes | NR | NR |
| Zheng, 2019 [31] | Sprague-Dawley | 5 weeks old | 16 weeks | 900 mg/kg/day | NR | NR | Yes | Yes |
| First Author, Year | Mouse Strain | Age | Duration of Treatment | Dosage of Treatment | Determinants of Adiposity | |||
|---|---|---|---|---|---|---|---|---|
| Decreased Adipocyte Size | Decreased Fat Mass | Decreased TG | Decreased TC | |||||
| Lee, 2017 [32] | Sprague-Dawley | 6 weeks old | 8 weeks and 12 weeks | 500 mg/kg/day | NR | Yes | Yes | NR |
| Bornstein, 2017 [24] | C57BL/6J | 18 weeks old | 6 weeks | 300 mg/kg/day | Yes | No (No Changes) | NR | NR |
| Kim, 2016 [33] | C57BL/6 | 4 weeks old | 14 weeks | 10 mg/kg and 50 mg/kg | Yes | NR | Yes | Yes |
| Luo, 2016 [34] | C57BL/6 | 8–10 weeks old | 4 weeks | 250 mg/kg/day | Yes | Yes | NR | NR |
| de Oliveira Santana, 2016 [35] | SWISS | 4 weeks old | 2 months | 100 mg/kg/day | Yes | NR | Yes | Yes |
| Ismail, 2013 [36] | Wistar | 4 weeks old | 2 weeks | 400 mg/kg/day | NR | NR | Yes | Yes |
| Pei, 2012 [37] | Sprague-Dawley | 4 weeks old | 6 weeks | 200 mg/kg/day | NR | Yes | NR | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, D.K.W.; Kadirvelu, A.; Pamidi, N. Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review. J. Clin. Med. 2022, 11, 4193. https://doi.org/10.3390/jcm11144193
Loh DKW, Kadirvelu A, Pamidi N. Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review. Journal of Clinical Medicine. 2022; 11(14):4193. https://doi.org/10.3390/jcm11144193
Chicago/Turabian StyleLoh, Darren Kin Wai, Amudha Kadirvelu, and Narendra Pamidi. 2022. "Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review" Journal of Clinical Medicine 11, no. 14: 4193. https://doi.org/10.3390/jcm11144193
APA StyleLoh, D. K. W., Kadirvelu, A., & Pamidi, N. (2022). Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review. Journal of Clinical Medicine, 11(14), 4193. https://doi.org/10.3390/jcm11144193