
Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis
Abstract
1. Introduction
2. Historic Background/Mechanism of Action/Molecular Effects
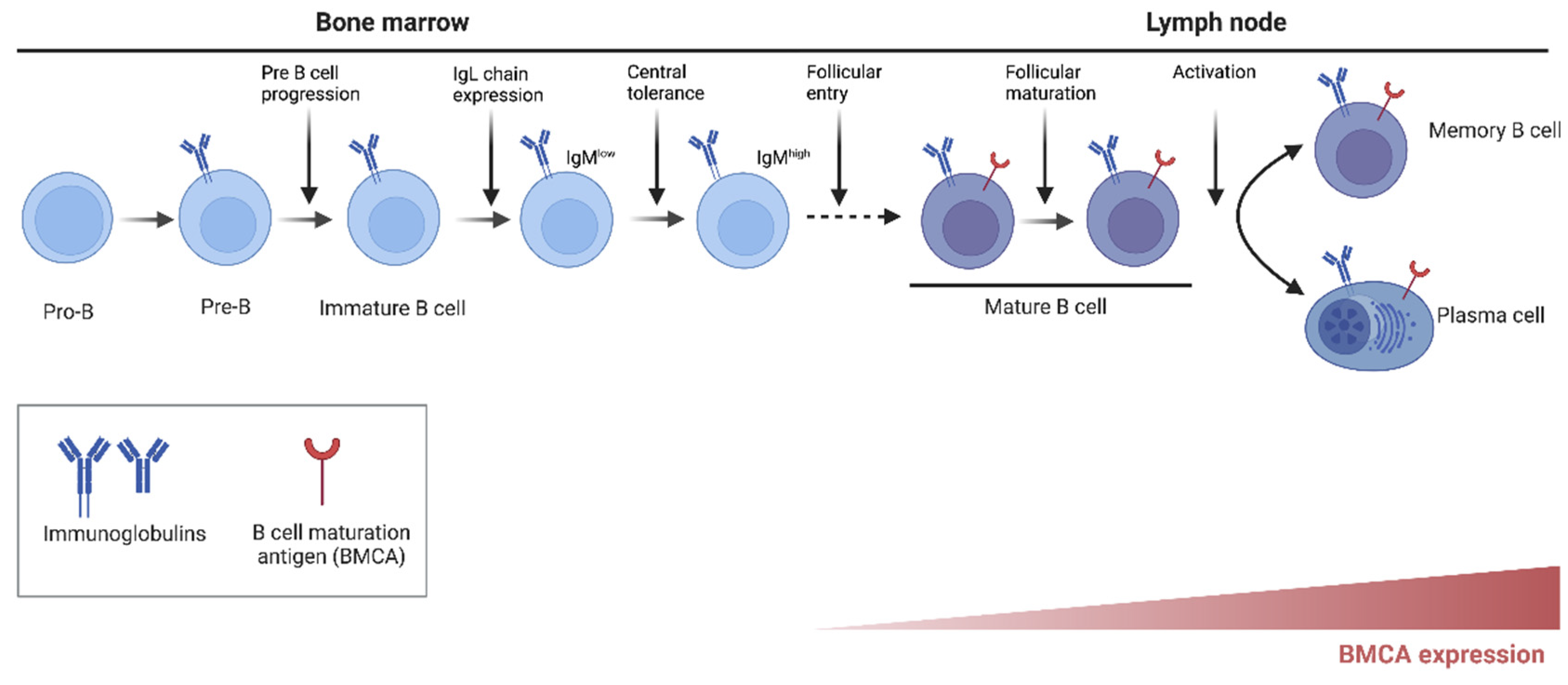
3. Role of BTK in B-Cell Development and Function


4. BTK in Other Myeloid Cells
5. BTK and T Lymphocytes
6. BTK and Microglia
7. Development and Current Use of BTK Inhibitors as Cancer Therapy
8. Role in Autoimmune Disorders
9. Systemic Lupus Erythematosus (SLE)
10. Rheumatoid Arthritis (RA)
11. Multiple Sclerosis (MS)
12. Presumed Mechanism of Action of BTKi in MS
12.1. Checkpoint 1. Antigen Presentation and Breakdown of Immune Tolerance
12.2. Checkpoint 2. CNS Migration of Autoreactive Cells
12.3. Checkpoint 3. Secondary Activation of the Autoreactive Cells within the CNS
12.4. Checkpoint 4. Astrocyte Modulation
12.5. Checkpoint 5. Demyelination and Remyelination
12.6. Checkpoint 6. Axonal Injury
13. Pre-Clinical Studies of BTKi in MS
14. Clinical Studies of BTKi in MS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BLNK | B-cell linker |
| PLC-γ2 | 1-phosphatidylinositol-4,5-biphosphate phosphodiesterase gamma-2 |
| IP3 | inositol 1,4,5-trisphosphate |
| DAG | diacylglycerol |
| PKC | protein kinase C |
References
- Szilveszter, K.P.; Nemeth, T.; Mocsai, A. Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. BTK Signaling in B Cell Differentiation and Autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, J.M.; Blomberg, K.E.; Valiaho, J.; Vargas, L.; Heinonen, J.E.; Berglof, A.; Mohamed, A.J.; Nore, B.F.; Vihinen, M.; Smith, C.I. Bruton’s tyrosine kinase: Cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol. Rev. 2005, 203, 200–215. [Google Scholar] [CrossRef]
- Lopez-Herrera, G.; Vargas-Hernandez, A.; Gonzalez-Serrano, M.E.; Berron-Ruiz, L.; Rodriguez-Alba, J.C.; Espinosa-Rosales, F.; Santos-Argumedo, L. Bruton’s tyrosine kinase—An integral protein of B cell development that also has an essential role in the innate immune system. J. Leukoc. Biol. 2014, 95, 243–250. [Google Scholar] [CrossRef]
- Rip, J.; de Bruijn, M.J.W.; Appelman, M.K.; Pal Singh, S.; Hendriks, R.W.; Corneth, O.B.J. Toll-Like Receptor Signaling Drives Btk-Mediated Autoimmune Disease. Front. Immunol. 2019, 10, 95. [Google Scholar] [CrossRef]
- Haselmayer, P.; Camps, M.; Liu-Bujalski, L.; Nguyen, N.; Morandi, F.; Head, J.; O’Mahony, A.; Zimmerli, S.C.; Bruns, L.; Bender, A.T.; et al. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J. Immunol. 2019, 202, 2888–2906. [Google Scholar] [CrossRef]
- Martin, E.; Aigrot, M.S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2020, 5, 123–133. [Google Scholar] [CrossRef]
- Keaney, J.; Gasser, J.; Gillet, G.; Scholz, D.; Kadiu, I. Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2019, 14, 448–461. [Google Scholar] [CrossRef]
- Geladaris, A.; Hausler, D.; Weber, M.S. Microglia: The Missing Link to Decipher and Therapeutically Control MS Progression? Int. J. Mol. Sci. 2021, 22, 3461. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Sacco, K.A.; Abraham, R.S. Consequences of B-cell-depleting therapy: Hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy 2018, 10, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Bruton, O.C. Agammaglobulinemia. Pediatrics 1952, 9, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarstrom, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Tsukada, S.; Rawlings, D.J.; Witte, O.N. Role of Bruton’s tyrosine kinase in immunodeficiency. Curr. Opin. Immunol. 1994, 6, 623–630. [Google Scholar] [CrossRef]
- Mano, H. Tec family of protein-tyrosine kinases: An overview of their structure and function. Cytokine Growth Factor Rev. 1999, 10, 267–280. [Google Scholar] [CrossRef]
- Melchers, F. Checkpoints that control B cell development. J. Clin. Investig. 2015, 125, 2203–2210. [Google Scholar] [CrossRef]
- Torke, S.; Weber, M.S. Inhibition of Bruton’s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Expert Opin. Investig. Drugs 2020, 29, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Middendorp, S.; Dingjan, G.M.; Maas, A.; Dahlenborg, K.; Hendriks, R.W. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol. 2003, 171, 5988–5996. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavare, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef]
- Anderson, J.S.; Teutsch, M.; Dong, Z.; Wortis, H.H. An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 10966–10971. [Google Scholar] [CrossRef]
- Contreras, C.M.; Halcomb, K.E.; Randle, L.; Hinman, R.M.; Gutierrez, T.; Clarke, S.H.; Satterthwaite, A.B. Btk regulates multiple stages in the development and survival of B-1 cells. Mol. Immunol. 2007, 44, 2719–2728. [Google Scholar] [CrossRef]
- Crofford, L.J.; Nyhoff, L.E.; Sheehan, J.H.; Kendall, P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. [Google Scholar] [CrossRef] [PubMed]
- de Gorter, D.J.; Beuling, E.A.; Kersseboom, R.; Middendorp, S.; van Gils, J.M.; Hendriks, R.W.; Pals, S.T.; Spaargaren, M. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 2007, 26, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Spaargaren, M.; Beuling, E.A.; Rurup, M.L.; Meijer, H.P.; Klok, M.D.; Middendorp, S.; Hendriks, R.W.; Pals, S.T. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J. Exp. Med. 2003, 198, 1539–1550. [Google Scholar] [CrossRef]
- Hata, D.; Kawakami, Y.; Inagaki, N.; Lantz, C.S.; Kitamura, T.; Khan, W.N.; Maeda-Yamamoto, M.; Miura, T.; Han, W.; Hartman, S.E.; et al. Involvement of Bruton’s tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 1998, 187, 1235–1247. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Puig Cano, A.; Hasija, M.; Xiao, H.; Smith, C.I.; Cybulsky, M.I. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J. Immunol. 2008, 181, 288–298. [Google Scholar] [CrossRef]
- Kawakami, Y.; Yao, L.; Miura, T.; Tsukada, S.; Witte, O.N.; Kawakami, T. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross-linking. Mol. Cell. Biol. 1994, 14, 5108–5113. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Koga, T.; Okamoto, K.; Sakaguchi, S.; Arai, K.; Yasuda, H.; Takai, T.; Kodama, T.; Morio, T.; Geha, R.S.; et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008, 132, 794–806. [Google Scholar] [CrossRef]
- Quek, L.S.; Bolen, J.; Watson, S.P. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 1998, 8, 1137–1140. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Correale, J. BTK inhibitors as potential therapies for multiple sclerosis. Lancet Neurol. 2021, 20, 689–691. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef]
- Voss, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Sriram, S. Role of glial cells in innate immunity and their role in CNS demyelination. J. Neuroimmunol. 2011, 239, 13–20. [Google Scholar] [CrossRef]
- Bhasin, M.; Wu, M.; Tsirka, S.E. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunol. 2007, 8, 10. [Google Scholar] [CrossRef]
- Glendenning, L.G.R.; Dufault, M.; Chretien, N.; Proto, J.; Zhang, M.; Lamorte, M.; Havari, E.; Turner, T.; Chomyk, A.; Christie, E.; et al. Decoding Bruton’s tyrosine kinase signalling in neuroinflammation. MSVirtual 2020, 26, 270. [Google Scholar]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Robak, E.; Robak, T. Bruton’s Kinase Inhibitors for the Treatment of Immunological Diseases: Current Status and Perspectives. J. Clin. Med. 2022, 11, 2807. [Google Scholar] [CrossRef] [PubMed]
- Zain, R.; Vihinen, M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front. Immunol. 2021, 12, 694853. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Wang, C.; Xu, Z.; Zhang, Y.; Liu, Y.; Zhao, G.; Ling, Y. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur. J. Med. Chem. 2022, 229, 114009. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef]
- Kim, H.-O. Development of BTK inhibitors for the treatment of B-cell malignancies. Arch. Pharmacal. Res. 2019, 42, 171–181. [Google Scholar] [CrossRef]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Deng, Y.; Chen, Y.; Yu, K.; Wang, A.; Liang, Q.; Wang, W.; Chen, C.; Wu, H.; Hu, C.; et al. Structure-activity relationship investigation for benzonaphthyridinone derivatives as novel potent Bruton’s tyrosine kinase (BTK) irreversible inhibitors. Eur. J. Med. Chem. 2017, 137, 545–557. [Google Scholar] [CrossRef]
- Ganatra, S.; Sharma, A.; Shah, S.; Chaudhry, G.M.; Martin, D.T.; Neilan, T.G.; Mahmood, S.S.; Barac, A.; Groarke, J.D.; Hayek, S.S.; et al. Ibrutinib-Associated Atrial Fibrillation. JACC Clin. Electrophysiol. 2018, 4, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G. Deciphering Ibrutinib Resistance in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1451–1452. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Doorduijn, J.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): A single-arm, multicentre, phase 2 trial. Lancet 2018, 391, 659–667. [Google Scholar] [CrossRef]
- Dhillon, S. Tirabrutinib: First Approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef]
- Walter, H.S.; Rule, S.A.; Dyer, M.J.; Karlin, L.; Jones, C.; Cazin, B.; Quittet, P.; Shah, N.; Hutchinson, C.V.; Honda, H.; et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016, 127, 411–419. [Google Scholar] [CrossRef]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Aalipour, A.; Advani, R.H. Bruton’s tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: Focus on ibrutinib. Ther. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.M.; Radsak, M.P.; Brunner, C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef] [PubMed]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef]
- Jansson, L.; Holmdahl, R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin. Exp. Immunol. 1993, 94, 459–465. [Google Scholar] [CrossRef]
- Mina-Osorio, P.; LaStant, J.; Keirstead, N.; Whittard, T.; Ayala, J.; Stefanova, S.; Garrido, R.; Dimaano, N.; Hilton, H.; Giron, M.; et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013, 65, 2380–2391. [Google Scholar] [CrossRef]
- Chang, B.Y.; Huang, M.M.; Francesco, M.; Chen, J.; Sokolove, J.; Magadala, P.; Robinson, W.H.; Buggy, J.J. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 2011, 13, R115. [Google Scholar] [CrossRef]
- Chalmers, S.A.; Wen, J.; Doerner, J.; Stock, A.; Cuda, C.M.; Makinde, H.M.; Perlman, H.; Bosanac, T.; Webb, D.; Nabozny, G.; et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res. Ther. 2018, 20, 10. [Google Scholar] [CrossRef]
- Kil, L.P.; de Bruijn, M.J.; van Nimwegen, M.; Corneth, O.B.; van Hamburg, J.P.; Dingjan, G.M.; Thaiss, F.; Rimmelzwaan, G.F.; Elewaut, D.; Delsing, D.; et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012, 119, 3744–3756. [Google Scholar] [CrossRef]
- Gabizon, R.; London, N. A Fast and Clean BTK Inhibitor. J. Med. Chem. 2020, 63, 5100–5101. [Google Scholar] [CrossRef]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Rankin, A.L.; Seth, N.; Keegan, S.; Andreyeva, T.; Cook, T.A.; Edmonds, J.; Mathialagan, N.; Benson, M.J.; Syed, J.; Zhan, Y.; et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 2013, 191, 4540–4550. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.; Vanarsa, K.; Bashmakov, A.; Grewal, S.; Sajitharan, D.; Chang, B.Y.; Buggy, J.J.; Zhou, X.J.; Du, Y.; Satterthwaite, A.B.; et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res. Ther. 2012, 14, R243. [Google Scholar] [CrossRef]
- Herman, A.E.; Chinn, L.W.; Kotwal, S.G.; Murray, E.R.; Zhao, R.; Florero, M.; Lin, A.; Moein, A.; Wang, R.; Bremer, M.; et al. Safety, Pharmacokinetics, and Pharmacodynamics in Healthy Volunteers Treated With GDC-0853, a Selective Reversible Bruton’s Tyrosine Kinase Inhibitor. Clin. Pharmacol. Ther. 2018, 103, 1020–1028. [Google Scholar] [CrossRef]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor Fenebrutinib (GDC-0853) in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef]
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) Affects Markers of B Cell Activation, Chemotaxis, and Osteoclasts in Patients with Rheumatoid Arthritis: Results from a Mechanistic Study. Rheumatol. Ther. 2020, 7, 101–119. [Google Scholar] [CrossRef]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A.; et al. Fenebrutinib versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial (ANDES Study). Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef]
- Li, R.; Tang, H.; Burns, J.C.; Hopkins, B.T.; Le Coz, C.; Zhang, B.; de Barcelos, I.P.; Romberg, N.; Goldstein, A.C.; Banwell, B.L.; et al. BTK inhibition limits B-cell-T-cell interaction through modulation of B-cell metabolism: Implications for multiple sclerosis therapy. Acta Neuropathol. 2022, 143, 505–521. [Google Scholar] [CrossRef]
- Torke, S.; Pretzsch, R.; Hausler, D.; Haselmayer, P.; Grenningloh, R.; Boschert, U.; Bruck, W.; Weber, M.S. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol. 2020, 140, 535–548. [Google Scholar] [CrossRef]
- Di Paolo, J.A.; Huang, T.; Balazs, M.; Barbosa, J.; Barck, K.H.; Bravo, B.J.; Carano, R.A.; Darrow, J.; Davies, D.R.; DeForge, L.E.; et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ling, L.; Qi, L.; Chong, Y.; Xue, L. Bruton’s Tyrosine Kinase (BTK) Inhibitor (Ibrutinib)-Suppressed Migration and Invasion of Prostate Cancer. Onco Targets Ther. 2020, 13, 4113–4122. [Google Scholar] [CrossRef] [PubMed]
- Tout, I.; Miossec, P. The role of B cells and their interactions with stromal cells in the context of inflammatory autoimmune diseases. Autoimmun. Rev. 2022, 21, 103098. [Google Scholar] [CrossRef] [PubMed]
- Alankus, Y.B.; Grenningloh, R.; Haselmayer, P.; Bender, A.T.; Bruttger, J. Inhibition of Bruton’s Tyrosine Kinase Prevents Inflammatory Macrophage Differentiation: A Potential Role in Multiple Sclerosis. In Proceedings of the Annual Meeting of the Consortium of Multiple Sclerosis Centers, Seattle, WA, USA, 28 May–1 June 2019. [Google Scholar]
- Sala, L.; Cirillo, G.; Riva, G.; Romano, G.; Giussani, C.; Cialdella, A.; Todisco, A.; Virtuoso, A.; Cerrito, M.G.; Bentivegna, A.; et al. Specific Expression of a New Bruton Tyrosine Kinase Isoform (p65BTK) in the Glioblastoma Gemistocytic Histotype. Front. Mol. Neurosci. 2019, 12, 2. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.Y.; Nam, Y.; Kang, H.J.; Cho, H.J.; Kim, J.; Hoe, H.S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflammation 2018, 15, 271. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Gruber, R.C.; Chretien, N.; Dufault, M.R.; Proto, J.; Zhang, M.; LaMorte, M.; Havari, E.; Samad, T.A.; Turner, T.; Chomyk, A.; et al. Central Effects of BTK Inhibition in Neuroinflammation (808). Neurology 2020, 94, 808. [Google Scholar]
- Crespo, O.; Kang, S.C.; Daneman, R.; Lindstrom, T.M.; Ho, P.P.; Sobel, R.A.; Steinman, L.; Robinson, W.H. Tyrosine kinase inhibitors ameliorate autoimmune encephalomyelitis in a mouse model of multiple sclerosis. J. Clin. Immunol. 2011, 31, 1010–1020. [Google Scholar] [CrossRef]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallstrom, E.; Zhang, X.; Mares, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Owens, T.D.; Smith, P.F.; Redfern, A.; Xing, Y.; Shu, J.; Karr, D.E.; Hartmann, S.; Francesco, M.R.; Langrish, C.L.; Nunn, P.A.; et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin. Transl. Sci. 2022, 15, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Bar-Or, A.; Francis, G.; Giovannoni, G.; Kappos, L.; Nicholas, J.; Oh, J.; Sormani, M.; Stoll, S.; Weber, M. Examination of fenebrutinib, a highly selective BTKi, on disease progression of multiple sclerosis. Mult. Scler. J. 2020, 26, 220. [Google Scholar]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Casteran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Benrabah, R.; Schmidt, N.; Zephir, H.; Clavelou, P.; Vongsouthi, C.; Dubreuil, P.; Moussy, A.; Hermine, O. Masitinib treatment in patients with progressive multiple sclerosis: A randomized pilot study. BMC Neurol. 2012, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Brieva-Ruiz, L.; Fox, R.J.; Paul, F.; Ramio-Torrenta, L.; Schwab, M.; Moussy, A.; Mansfield, C.; Hermine, O.; Maciejowski, M. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis: A Randomized, Phase 3, Clinical Trial. Neurol.-Neuroimmunol. Neuroinflamm. 2022, 9, e1148. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Drug | BTK Binding | Phase II Studies | Phase III Studies |
|---|---|---|---|
| Evobrutinib | Covalent, irreversible | RMS and active SPMS, reduced enhancing lesions compared to placebo | RMS (evolutionRMS1 and RMS2), evobrutinib vs. teriflunomide (NCT04338022, NCT04338061) |
| Tolebrutinib | Covalent, irreversible | RRMS and relapsing SPMS, reduced new active brain MRI lesions | RMS, tolebrutinib vs. teriflunomide PPMS and non-relapsing SPMS, compared to placebo (NCT04411641, NCT04458051, NCT04410991, and NCT04410978) |
| Fenebrutinib | Non-covalent, highly selective, reversible | Relapsing MS and active SPMS, fenebrutinib vs. teriflunomide Primary progressive MS, fenebrutinib vs. ocrelizumab (NCT04586023, NCT04586010, NCT04544449) | |
| Remibrutinib | Covalent irreversible | RMS, remibrutinib vs. teriflunomide (NCT05147220, NCT05156281) | |
| Orelabrutinib | Covalent, irreversible | Randomized, double-blind, placebo-controlled study ongoing (NCT04711148) | |
| Masitinib | Selective tyrosine kinase inhibitor | Randomized, double-blind, 2 parallel-group, placebo-controlled trial in PPMS and non-relapsing SPMS showed positive effect on disability progression A confirmatory phase 3 study ongoing, PPMS or non-relapsing SPMS, compared to placebo (NCT05441488) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, N.; Padron, E.J.; Rammohan, K.W.; Goodman, C.F. Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis. J. Clin. Med. 2022, 11, 6139. https://doi.org/10.3390/jcm11206139
Garg N, Padron EJ, Rammohan KW, Goodman CF. Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis. Journal of Clinical Medicine. 2022; 11(20):6139. https://doi.org/10.3390/jcm11206139
Chicago/Turabian StyleGarg, Neeta, Elizabeth Jordan Padron, Kottil W. Rammohan, and Courtney Frances Goodman. 2022. "Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis" Journal of Clinical Medicine 11, no. 20: 6139. https://doi.org/10.3390/jcm11206139
APA StyleGarg, N., Padron, E. J., Rammohan, K. W., & Goodman, C. F. (2022). Bruton’s Tyrosine Kinase Inhibitors: The Next Frontier of B-Cell-Targeted Therapies for Cancer, Autoimmune Disorders, and Multiple Sclerosis. Journal of Clinical Medicine, 11(20), 6139. https://doi.org/10.3390/jcm11206139