
Retrospective Analysis of Prognostic Factors in Pediatric Patients with Adrenocortical Tumor from Unique Tertiary Center with Long-Term Follow-Up
 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Epidemiologic Features: Age, Gender, and Geographic Origin from Brazil
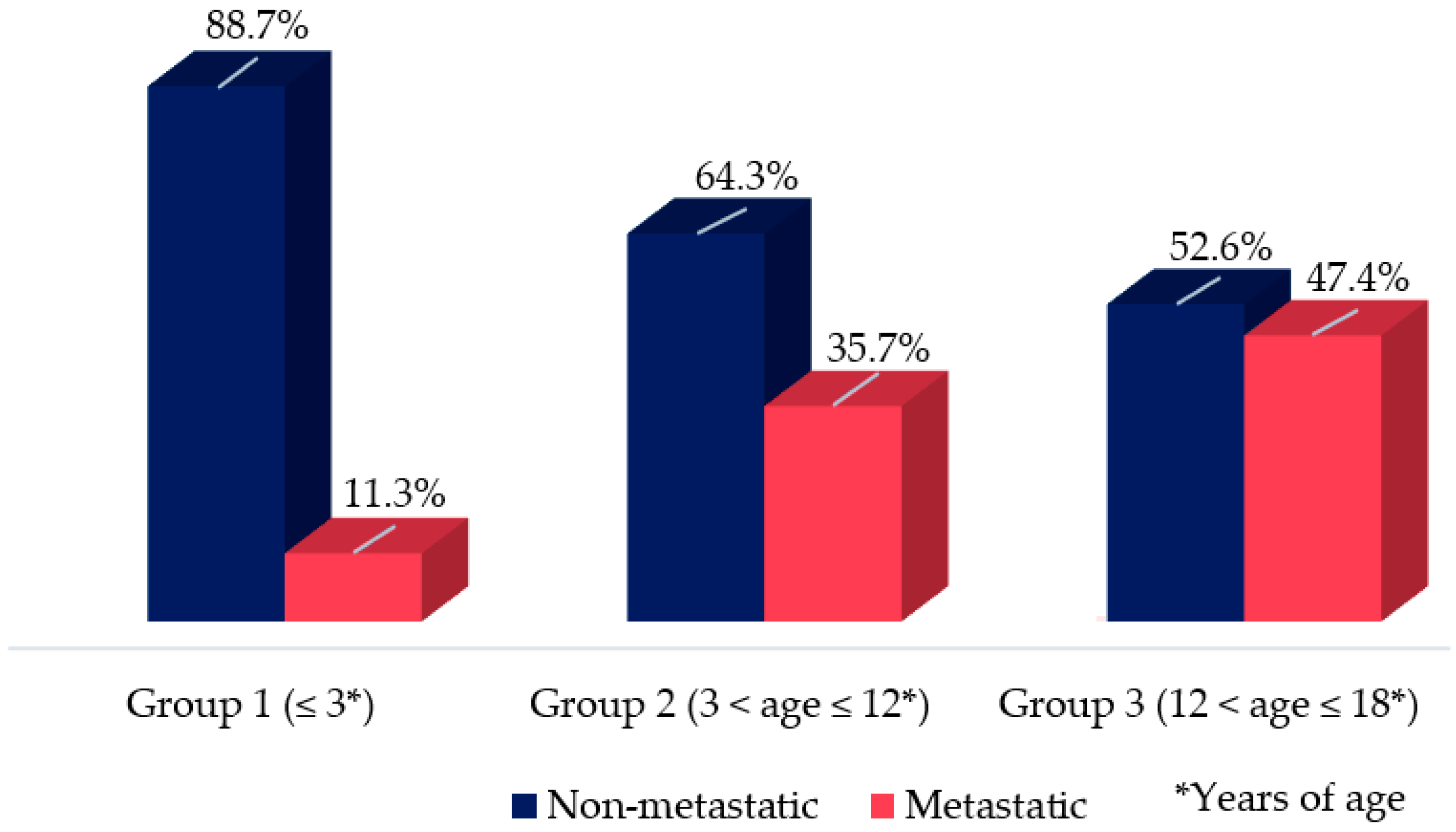
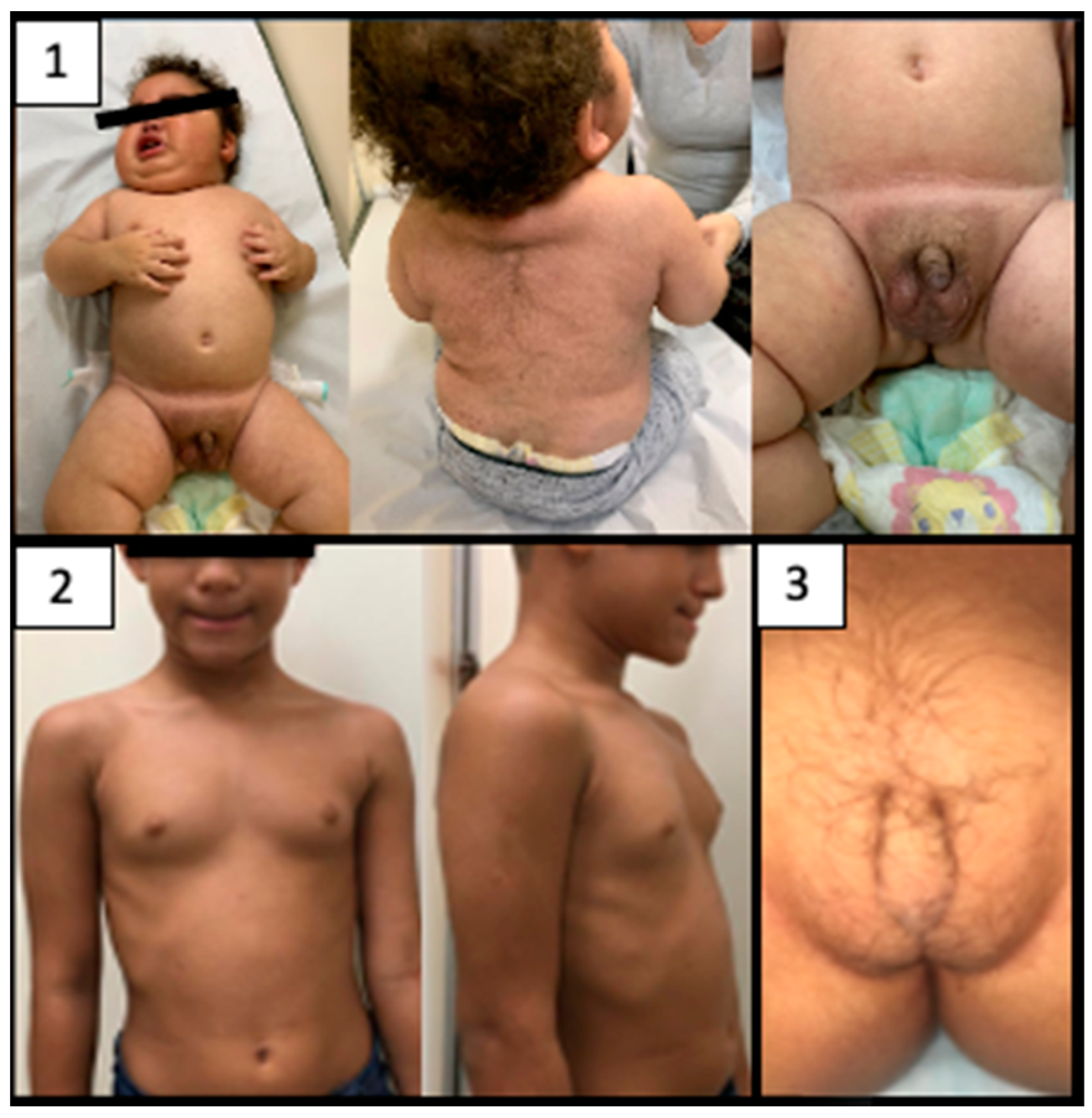
3.2. Clinical Presentation
3.3. Hormonal Data
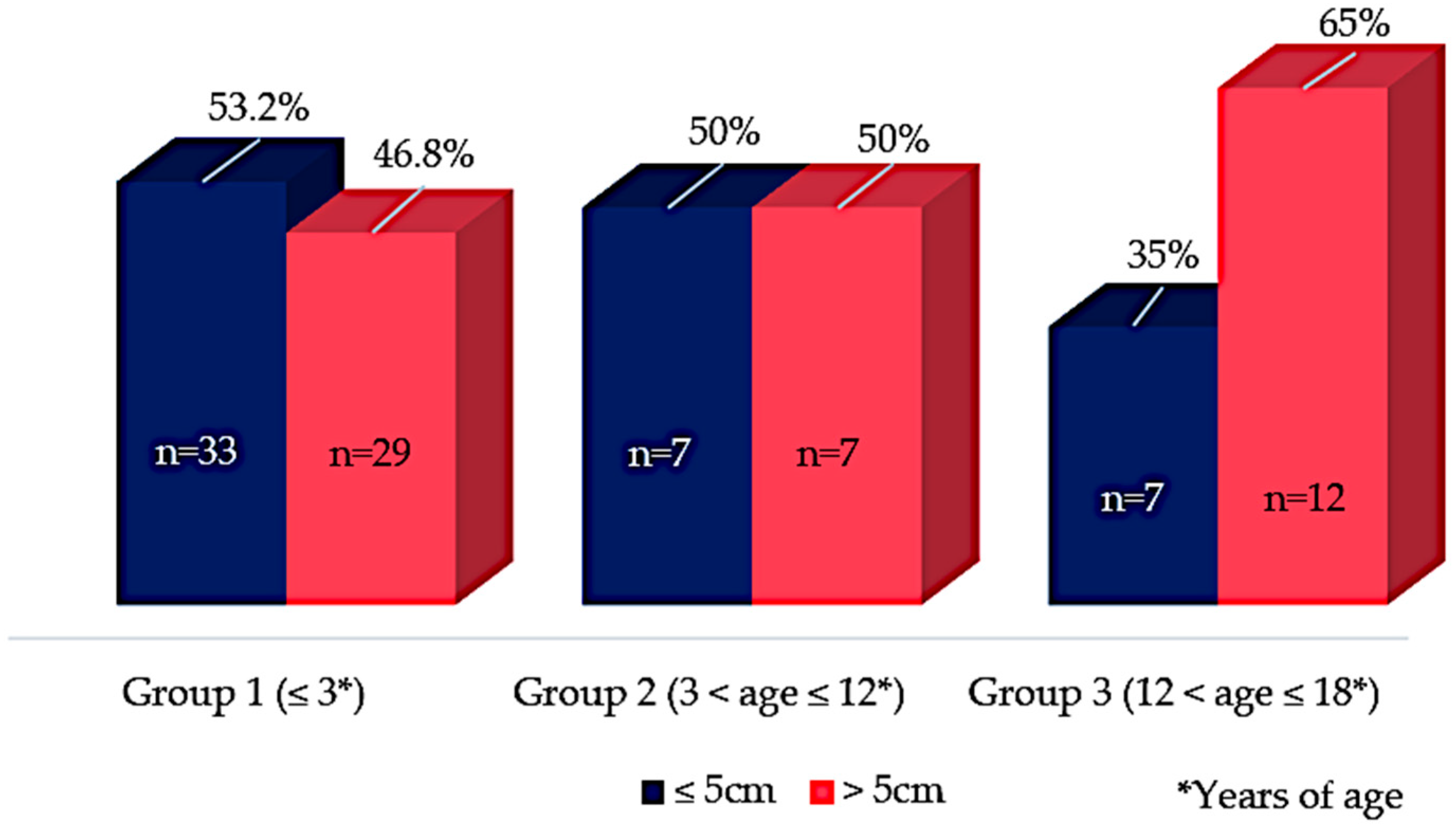
3.4. Radiological Data
3.5. Histological Data (Weiss/Modified Weiss Score and Wieneke Index) and Staging (MacFarlane and TNM)
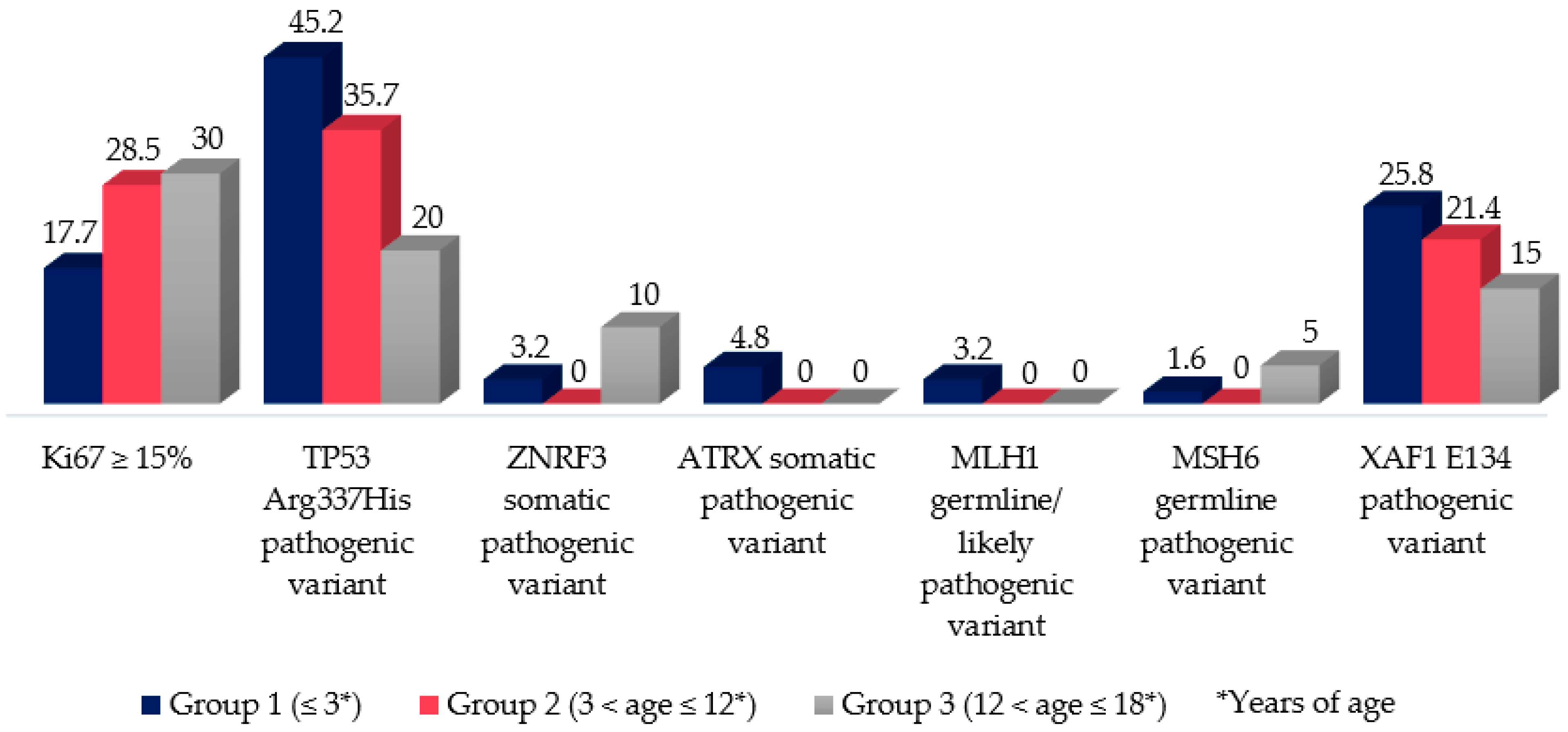
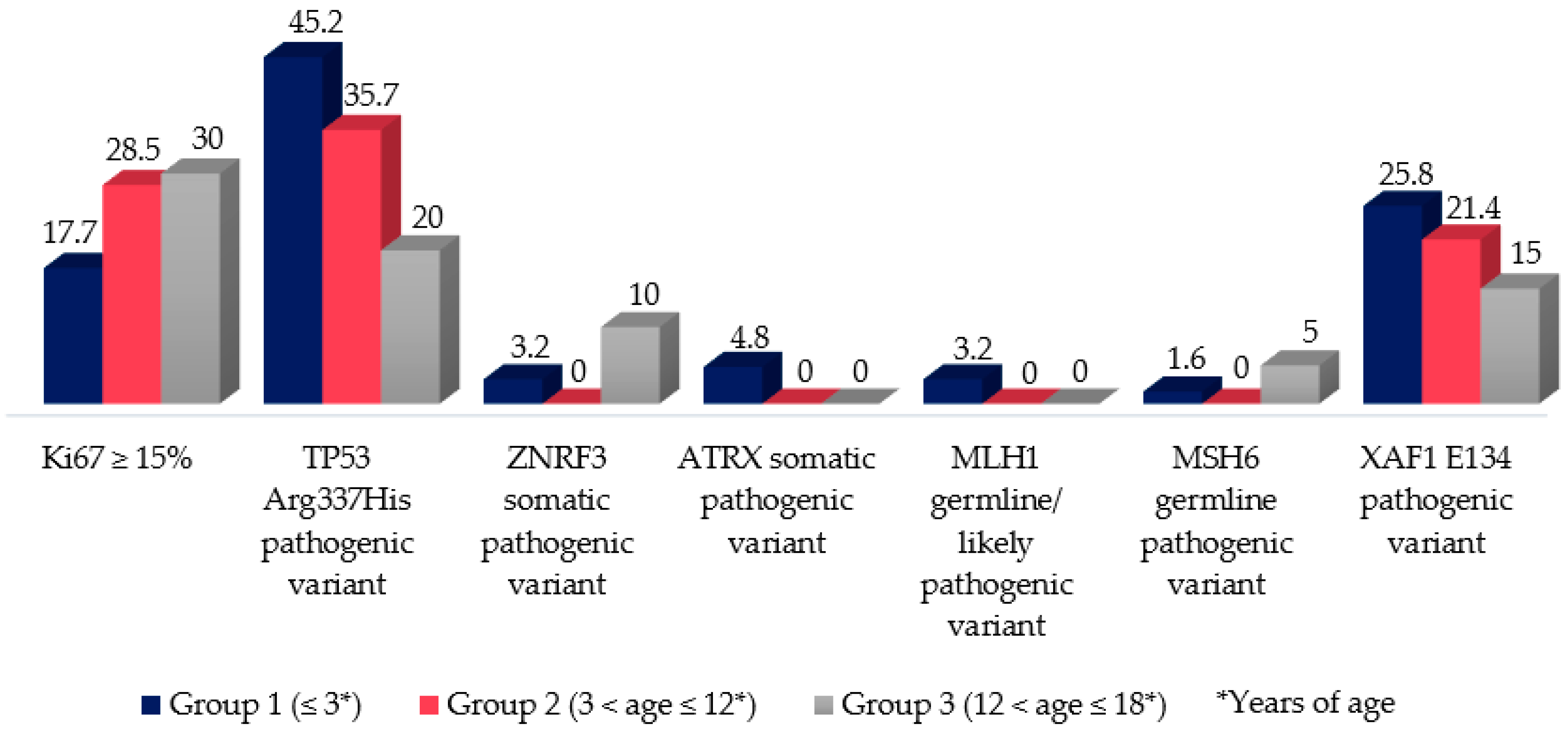
3.6. Pediatric Adrenocortical Tumors Molecular Markers (Melan A, SF1, Inhibin, p53, and Ki67)
3.7. Genetic Analysis (TP53, BUB1B-PINK1, IGF-IR, ATRX, ZNRF3, MLH1, MSH6, and XAF1): Compiled Data from Previously Published Manuscript of This Cohort
3.8. Treatment
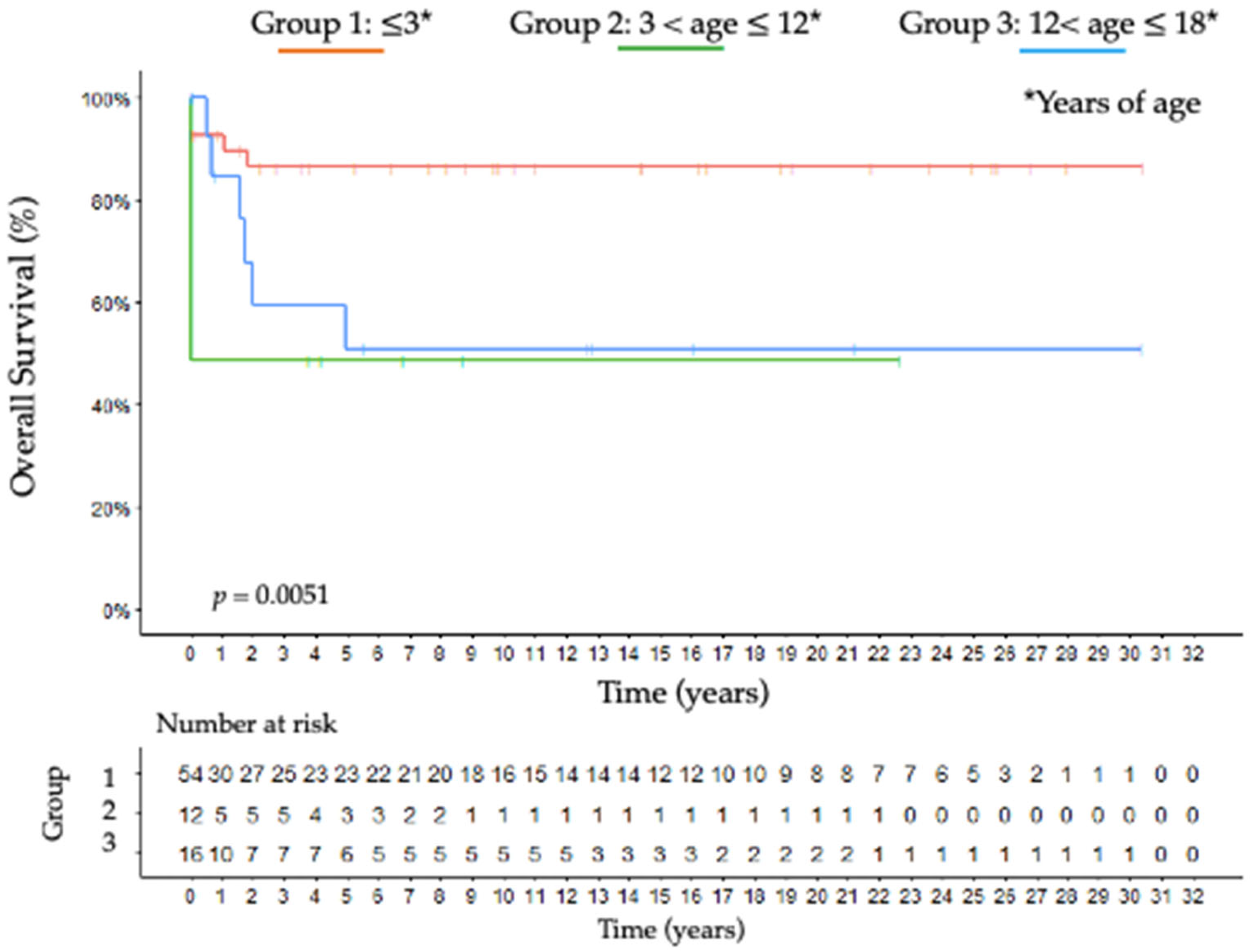
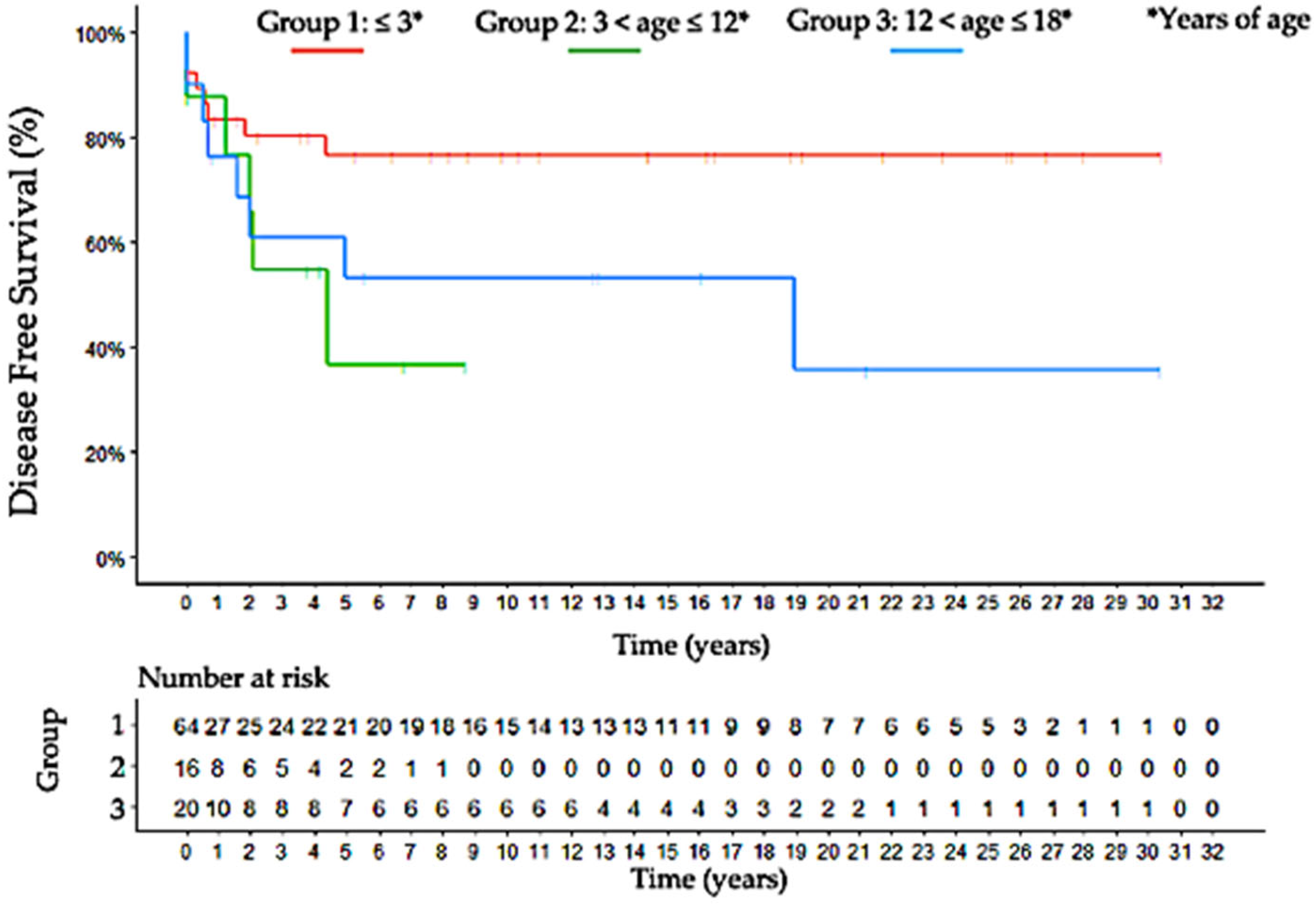
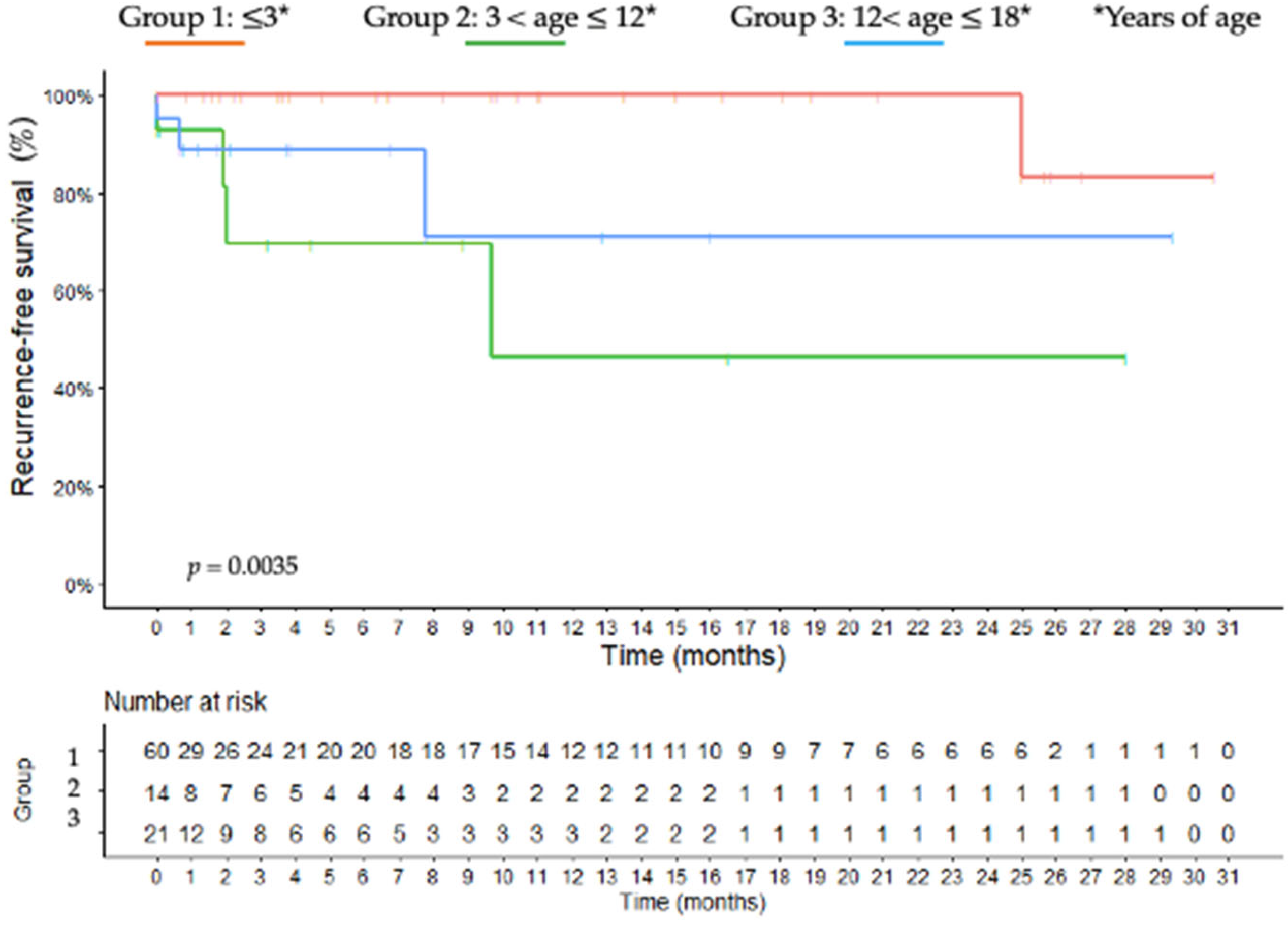
3.9. Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wajchenberg, B.L.; Albergaria Pereira, M.A.; Medonca, B.B.; Latronico, A.C.; Campos Carneiro, P.; Alves, V.A.; Zerbini, M.C.; Liberman, B.; Carlos Gomes, G.; Kirschner, M.A. Adrenocortical carcinoma: Clinical and laboratory observations. Cancer 2000, 88, 711–736. [Google Scholar] [CrossRef]
- McAteer, J.P.; Huaco, J.A.; Gow, K.W. Predictors of survival in pediatric adrenocortical carcinoma: A Surveillance, Epidemiology, and End Results (SEER) program study. J. Pediatr. Surg. 2013, 48, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, B.B.; Lucon, A.M.; Menezes, C.A.; Saldanha, L.B.; Latronico, A.C.; Zerbini, C.; Madureira, G.; Domenice, S.; Albergaria, M.A.; Camargo, M.H. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J. Urol. 1995, 154, 2004–2009. [Google Scholar] [CrossRef]
- Sbiera, S.; Schmull, S.; Assie, G.; Voelker, H.U.; Kraus, L.; Beyer, M.; Ragazzon, B.; Beuschlein, F.; Willenberg, H.S.; Hahner, S.; et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J. Clin. Endocrinol. Metab. 2010, 95, E161–E171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pianovski, M.A.; Maluf, E.M.; de Carvalho, D.S.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Boffetta, P.; Zancanella, P.; Figueiredo, B.C. Mortality rate of adrenocortical tumors in children under 15 years of age in Curitiba, Brazil. Pediatr. Blood Cancer 2006, 47, 56–60. [Google Scholar] [CrossRef]
- Pereira, R.M.; Michalkiewicz, E.; Sandrini, F.; Figueiredo, B.C.; Pianovski, M.; França, S.N.; Boguszewski, M.C.; Costa, O.; Cat, I.; Lacerda Filho, L.; et al. Childhood adrenocortical tumors. Arq. Bras. Endocrinol. Metabol. 2004, 48, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.A.G.; Melbert, D.; Krapcho, M.; Stinchcomb, D.G.; Howlader, N.; Horner, M.J.; Mariotto, A.; Miller, B.A.; Feuer, E.J.; Altekruse, S.F.; et al. SEER Cancer Statistics Review, 1975–2005. National Cancer Institute. Available online: https://seer.cancer.gov/csr/1975_2005/ (accessed on 12 April 2022).
- Bernstein, L.; Gurney, J.G. Carcinomas and Other Malignant Epithelial Neoplasms. In Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; Ries, L.A.G., Smith, M.A., Gurney, J.G., Linet, M., Tamra, T., Young, J.L., Bunin, G.R., Eds.; National Cancer Institute, SEER Program: Bethesda, MD, USA, 1999. [Google Scholar]
- Brondani, V.B.; Montenegro, L.; Lacombe, A.M.F.; Magalhães, B.M.; Nishi, M.Y.; Funari, M.F.A.; Narcizo, A.M.; Cardoso, L.C.; Siqueira, S.A.C.; Zerbini, M.C.N.; et al. High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers 2020, 12, 621. [Google Scholar] [CrossRef] [Green Version]
- Renaux-Petel, M.; Charbonnier, F.; Théry, J.C.; Fermey, P.; Lienard, G.; Bou, J.; Coutant, S.; Vezain, M.; Kasper, E.; Fourneaux, S.; et al. Contribution of de novo and mosaic. J. Med. Genet. 2018, 55, 173–180. [Google Scholar] [CrossRef]
- Giacomazzi, J.; Selistre, S.G.; Rossi, C.; Alemar, B.; Santos-Silva, P.; Pereira, F.S.; Netto, C.B.; Cossio, S.L.; Roth, D.E.; Brunetto, A.L.; et al. Li-Fraumeni and Li-Fraumeni-like syndrome among children diagnosed with pediatric cancer in Southern Brazil. Cancer 2013, 119, 4341–4349. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Brondani, V.B.; Helena, V.P.; Charchar, H.L.S.; Zerbini, M.C.N.; Leite, L.A.S.; Hoff, A.O.; Latronico, A.C.; Mendonca, B.B.; Diz, M.D.P.E.; et al. Clinical spectrum of Li-Fraumeni syndrome/Li-Fraumeni-like syndrome in Brazilian individuals with the TP53 p.R337H mutation. J. Steroid Biochem. Mol. Biol. 2019, 190, 250–255. [Google Scholar] [CrossRef]
- Pinto, E.M.; Figueiredo, B.C.; Chen, W.; Galvao, H.C.R.; Formiga, M.N.; Fragoso, M.C.B.V.; Ashton-Prolla, P.; Ribeiro, E.M.S.F.; Felix, G.; Costa, T.E.B.; et al. XAF1 as a modifier of p53 function and cancer susceptibility. Sci. Adv. 2020, 6, eaba3231. [Google Scholar] [CrossRef]
- Brondani, V.B.; Lacombe, A.M.F.; Mariani, B.M.P.; Montenegro, L.; Soares, I.C.; Bezerra-Neto, J.E.; Tanno, F.Y.; Srougi, V.; Chambo, J.L.; Mendonca, B.B.; et al. Low Protein Expression of both ATRX and ZNRF3 as Novel Negative Prognostic Markers of Adult Adrenocortical Carcinoma. Int. J. Mol. Sci. 2021, 22, 1238. [Google Scholar] [CrossRef]
- de Reyniès, A.; Assié, G.; Rickman, D.S.; Tissier, F.; Groussin, L.; René-Corail, F.; Dousset, B.; Bertagna, X.; Clauser, E.; Bertherat, J. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 2009, 27, 1108–1115. [Google Scholar] [CrossRef]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.; Nussenbaum, B.; Wang, E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Martins-Filho, S.N.; Almeida, M.Q.; Soares, I.; Wakamatsu, A.; Alves, V.A.F.; Fragoso, M.C.B.V.; Zerbini, M.C.N. Clinical Impact of Pathological Features Including the Ki-67 Labeling Index on Diagnosis and Prognosis of Adult and Pediatric Adrenocortical Tumors. Endocr. Pathol. 2021, 32, 288–300. [Google Scholar] [CrossRef]
- Wang, H.; Li, G. Extreme learning machine Cox model for high-dimensional survival analysis. Stat. Med. 2019, 38, 2139–2156. [Google Scholar] [CrossRef]
- Wileyto, E.P.; Li, Y.; Chen, J.; Heitjan, D.F. Assessing the fit of parametric cure models. Biostatistics 2013, 14, 340–350. [Google Scholar] [CrossRef] [Green Version]
- Araujo-Castro, M.; Sampedro Núñez, M.A.; Marazuela, M. Autonomous cortisol secretion in adrenal incidentalomas. Endocrine 2019, 64, 1–13. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Fragoso, M.C.; Lotfi, C.F.; Santos, M.G.; Nishi, M.Y.; Costa, M.H.; Lerario, A.M.; Maciel, C.C.; Mattos, G.E.; Jorge, A.A.; et al. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J. Clin. Endocrinol. Metab. 2008, 93, 3524–3531. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.I.; Suartz, C.V.; Neto, R.P.; Berjeaut, R.H.; Mendonca, B.; Almeida, M.Q.; Fragoso, M.C.V.; Dénes, F.T. Management of functioning pediatric adrenal tumors. J. Pediatr. Surg. 2021, 56, 768–771. [Google Scholar] [CrossRef]
- Zambaiti, E.; Duci, M.; De Corti, F.; Gamba, P.; Dall’Igna, P.; Ghidini, F.; Virgone, C. Clinical prognostic factors in pediatric adrenocortical tumors: A meta-analysis. Pediatr. Blood Cancer 2021, 68, e28836. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.M.; Faucz, F.R.; Paza, L.Z.; Wu, G.; Fernandes, E.S.; Bertherat, J.; Stratakis, C.A.; Lalli, E.; Ribeiro, R.C.; Rodriguez-Galindo, C.; et al. Germline Variants in Phosphodiesterase Genes and Genetic Predisposition to Pediatric Adrenocortical Tumors. Cancers 2020, 12, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso, M.C.; Almeida, M.Q.; Mazzuco, T.L.; Mariani, B.M.; Brito, L.P.; Gonçalves, T.C.; Alencar, G.A.; Lima, L.e.O.; Faria, A.M.; Bourdeau, I.; et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: Validation in a Brazilian cohort of adult and pediatric patients. Eur. J. Endocrinol. 2012, 166, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicquel, C.; Bertagna, X.; Gaston, V.; Coste, J.; Louvel, A.; Baudin, E.; Bertherat, J.; Chapuis, Y.; Duclos, J.M.; Schlumberger, M.; et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res. 2001, 61, 6762–6767. [Google Scholar] [PubMed]
- Brondani, V.B.; Fragoso, M.C.B.V. Pediatric adrenocortical tumor—Review and management update. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 177–186. [Google Scholar] [CrossRef]
- Faria, A.M.; Almeida, M.Q. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol. Cell Endocrinol. 2012, 351, 52–57. [Google Scholar] [CrossRef]
- Lalli, E.; Figueiredo, B.C. Pediatric adrenocortical tumors: What they can tell us on adrenal development and comparison with adult adrenal tumors. Front. Endocrinol. 2015, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Riedmeier, M.; Decarolis, B.; Haubitz, I.; Müller, S.; Uttinger, K.; Börner, K.; Reibetanz, J.; Wiegering, A.; Härtel, C.; Schlegel, P.G.; et al. Adrenocortical Carcinoma in Childhood: A Systematic Review. Cancers 2021, 13, 5266. [Google Scholar] [CrossRef]
- Evanoff, J.D.; Patel, S.G.; Hickey, K.J.; Rensing, A.J. Survival characteristics of localized pediatric adrenocortical carcinoma managed with adenectomy: A national cancer center database analysis. J. Pediatr. Urol. 2021, 17, 735.e1–735.e6. [Google Scholar] [CrossRef]
- Ribeiro, R.C.; Michalkiewicz, E.L.; Figueiredo, B.C.; DeLacerda, L.; Sandrini, F.; Pianovsky, M.D.; Sampaio, G.; Sandrini, R. Adrenocortical tumors in children. Braz. J. Med. Biol. Res. 2000, 33, 1225–1234. [Google Scholar] [CrossRef]
- Michalkiewicz, E.; Sandrini, R.; Figueiredo, B.; Miranda, E.C.; Caran, E.; Oliveira-Filho, A.G.; Marques, R.; Pianovski, M.A.; Lacerda, L.; Cristofani, L.M.; et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J. Clin. Oncol. 2004, 22, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, G.; Sun, H.; Li, K.; Dong, K.; Ma, Y.; Zheng, S. Clinical characteristics and prognosis of adrenocortical tumors in children. Pediatr. Surg. Int. 2019, 35, 365–371. [Google Scholar] [CrossRef]
- Wieneke, J.A.; Thompson, L.D.; Heffess, C.S. Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients. Am. J. Surg. Pathol. 2003, 27, 867–881. [Google Scholar] [CrossRef]
- Sandru, F.; Petca, R.C.; Carsote, M.; Petca, A.; Dumitrascu, M.C.; Ghemigian, A. Adrenocortical carcinoma: Pediatric aspects (Review). Exp. Ther. Med. 2022, 23, 287. [Google Scholar] [CrossRef]
- Kerkhofs, T.M.; Ettaieb, M.H.; Verhoeven, R.H.; Kaspers, G.J.; Tissing, W.J.; Loeffen, J.; Van den Heuvel-Eibrink, M.M.; De Krijger, R.R.; Haak, H.R. Adrenocortical carcinoma in children: First population-based clinicopathological study with long-term follow-up. Oncol. Rep. 2014, 32, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, D.A. Cancer of the adrenal cortex; the natural history, prognosis and treatment in a study of fifty-five cases. Ann. R Coll. Surg. Engl. 1958, 23, 155–186. [Google Scholar]
- Goldblum, J.R.; Shannon, R.; Kaldjian, E.P.; Thiny, M.; Davenport, R.; Thompson, N.; Lloyd, R.V. Immunohistochemical assessment of proliferative activity in adrenocortical neoplasms. Mod. Pathol. 1993, 6, 663–668. [Google Scholar]
- Stojadinovic, A.; Brennan, M.F.; Hoos, A.; Omeroglu, A.; Leung, D.H.; Dudas, M.E.; Nissan, A.; Cordon-Cardo, C.; Ghossein, R.A. Adrenocortical adenoma and carcinoma: Histopathological and molecular comparative analysis. Mod. Pathol. 2003, 16, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.; Boileau, M.; Hodges, C.V. Adrenal cortical carcinoma. J. Urol. 1978, 120, 660–665. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | No of Patients n (%) | Mean Age at Diagnosis (Years of Age) | Female (%) | Male (%) | From South/Southeast Regions of Brazil (n) | From Midwest Region of Brazil (n) | From Northeast Region of Brazil (n) | From Bolivia and United States (n) |
|---|---|---|---|---|---|---|---|---|
| 1 | 62 (68.4) | 1.9 | 45 | 17 | 46 | 10 | 5 | 1 |
| 2 | 14 (16.8) | 7 | 10 | 4 | 12 | 1 | 0 | 1 |
| 3 | 20 (21) | 17.3 | 17 | 3 | 12 | 3 | 3 | 0 |
| p = 0.03 (>3 years of age) | p = 0.09 | |||||||
| HR: 2.5; CI 95% 1.07–5.8 |
| Group | 11-Deoxycortisol (ng/mL) | 17-OHP (ng/mL) | SDHEA (ng/mL) | Testosterone (ng/dL) | Free Testosterone (pmol/L) | Estradiol (pg/mL) | LH (UI/L) | Cortisol (µg/dL) |
|---|---|---|---|---|---|---|---|---|
| 1 | 6 ± 8 (<0.5) | 15.3 ± 54.6 (<0.86) | 23614.2 ± 46241.2 (<1240) | 408.4 ± 775 (female: <14; male: <14) | 194.4 ± 266.5 (female: 2.4–37; male: 131–640) | 31.55 ± 52.2 (female: <21; male: <20) | 0.81 ± 1.88 (female: 2.4–12.6; male: 1.7–8.6) | 17.45 ± 53.48 (in the morning: 6.7–22.6) |
| 2 | 8 ± 7.9 (<0.5) | 3.2 ± 1.8 (<0.86) | 7429.5 ± 9264.7 (<852) | 363.6 ± 267.8 (female: <14; male: 3–32) | 193 ± 172.5 (female: 2.4–37; male: 131–640) | 27 ± 8.75 (female: 6–27; male: 25.8–60.7) | 0.23 ± 0.21 (female: 2.4–12.6; male: 1.7–8.6) | 12.96 ± 7.79 (in the morning: 6.7–22.6) |
| 3 | 257.3 ± 392.9 (<45) | 3.7 ± 3.3 (0.6–3.3) | 5834.3 ± 8493.2 (678–4127) | 395.4 ± 267.8 (female: <98; male: 200–830) | 132.7 ± 183.6 (female: 2.4–37; male: 131–640) | 45.81 ± 51.52 (female: follicular phase: 19–247; luteal phase 22–256; male: <56) | 4.17 ± 9.93 (female, follicular phase: 20–125; luteal phase: 5–17; male: 1.4–9.2) | 18.98 ± 4.85 (in the morning: 4–22) |
| p < 0.001 | p = 0.297 | p = 0.416 | p = 0.518 | p = 0.511 | p = 0.501 | p = 0.477 | p = 0.263 | |
| HR: 2.9 CI 95% 0.9–1.2 |
| Patients | Gender | Age (yrs) | Clinical | Tu Size (cm) | Weight (g) | Wieneke Index | Weiss Score | M. Weiss Score | Ki67 (%) | p53 p.R337H | MacF. Stage | 11-Deoxycortisol (<0.5 ng/mL) | Decease |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p = 0.09 | p = 0.03 (>3 yrs of age) | p = 0.057 | p < 0.05 (≥5 cm) | p < 0.001 (≥200 g) | p < 0.001 (≥3) | p = 0.002 (≥5) | p < 0.001 (≥5) | p < 0.05 (≥15%) | p > 0.05 | p < 0.001 (I–III) | p < 0.001 | ||
| HR: 2.5; CI 95% 1.07–5.8 | HR 0.042; CI 95% 0.002–1.1 | HR: 5.3; CI 95% 1.03–5.4 | HR: 16.2; CI 95% 0.9–2.1; | HR: 25.8; CI 95% 1.4–3.4 | HR: 6.2; CI 95% 1.2–8.3 | HR: 6.2; CI 95% 1.2–8.3 | HR: 8.1; CI 95% 1.2–8.2 | HR: 5.1; CI 95% 1–25.9 | HR: 2.9; CI 95% 0.9–1.2 | ||||
| #1 | F | 0.1 | V + C | 3.9 | 31.4 | 1 | 7 | 5 | 50 | yes | I | 3.17 | No |
| #2 | F | 0.1 | V | 6.4 | 47 | 3 | 8 | 6 | 30 | yes | II | 0.4 | No |
| #3 | M | 0.7 | V | 4 | n.a. | 3 | 6 | 6 | 43 | yes | I | n.a. | No |
| #4 | F | 0.8 | V | 4 | 25 | 3 | 6 | 5 | 40 | yes | IV | n.a. | No |
| #5 | F | 0.11 | V | 4.5 | 31.3 | 1 | 3 | 3 | 13 | no | I | 1.8 | No |
| #6 | F | 0.11 | V | 6.5 | 112 | 3 | 6 | 6 | 16 | yes | II | 6.6 | No |
| #7 | F | 0.11 | V + C | 8.5 | 138 | 3 | 4 | 3 | n.a. | yes | II | n.a. | No |
| #8 | F | 1.11 | V | 5 | 45 | 1 | 2 | 1 | 11 | no | I | n.a. | No |
| #9 | F | 0.8 | V + C | 3.5 | 50 | 2 | 6 | 6 | 31 | yes | I | n.a. | No |
| #10 | M | 0.9 | V + C | 8.5 | 89 | 4 | 9 | 7 | 10 | yes | III | 2.2 | Yes |
| #11 | F | 1 | V + C | 4.5 | n.a. | 1 | 1 | 2 | n.a. | no | I | n.a. | No |
| #12 | M | 1 | V + C | 11 | n.a. | 3 | 5 | 4 | n.a. | no | II | n.a. | No |
| #13 | F | 1 | V | 4.5 | 20 | n.a. | 2 | n.a. | n.a. | no | I | n.a. | No |
| #14 | F | 1 | V + C | 6 | 60 | n.a. | n.a. | n.a. | n.a. | no | III | n.a. | No |
| #15 | F | 1 | V + C | 6.4 | 60 | 1 | 2 | 2 | 15 | yes | II | n.a. | No |
| #16 | F | 1 | V | 3.3 | 11.8 | n.a. | 2 | n.a. | n.a. | yes | I | n.a. | No |
| #17 | F | 1 | V | 7 | 100 | 1 | 1 | 1 | n.a. | yes | II | n.a. | No |
| #18 | F | 1.1 | V | 5 | 190 | 2 | 6 | 4 | 1 | no | III | n.a. | No |
| #19 | M | 1.1 | V | 6 | n.a. | 3 | 6 | 5 | 31 | no | III | n.a. | No |
| #20 | F | 1.3 | V + C | 3.7 | 10 | 2 | 5 | 5 | 6 | yes | I | n.a. | No |
| #21 | F | 1.3 | V | 5 | 40 | 1 | 1 | 1 | n.a. | no | I | n.a. | No |
| #22 | M | 1.4 | V + C | 5 | 40 | 1 | 7 | 2 | 16 | yes | I | n.a. | No |
| #23 | M | 1.4 | V | 5 | 30 | 1 | 2 | 1 | 2 | yes | I | n.a. | No |
| #24 | M | 1.7 | V | 1.5 | 4 | 2 | 5 | 5 | 1 | yes | I | n.a. | No |
| #25 | F | 1.8 | V | 9.5 | 140 | 2 | 5 | 5 | 4 | no | II | n.a. | No |
| #26 | F | 1.9 | V | 3 | 10 | 3 | 6 | 6 | 2 | no | I | n.a. | No |
| #27 | M | 1.9 | V | 5.5 | 55 | 2 | 4 | 1 | 1 | no | II | n.a. | No |
| #28 | F | 1.9 | V + C | 7.5 | 38 | 2 | 5 | 1 | n.a. | yes | III | 3.9 | No |
| #29 | M | 2 | V | 3.5 | 10 | 4 | 4 | 5 | 11 | yes | I | n.a. | Yes |
| #30 | M | 2 | V + C | 12 | 250 | 6 | 7 | 6 | 19 | yes | IV | n.a. | Yes |
| #31 | F | 2 | V | 3 | n.a. | 1 | 1 | 1 | n.a. | yes | I | n.a. | Yes |
| #32 | F | 2.1 | n.a. | 7 | n.a. | 5 | 8 | 7 | n.a. | yes | II | n.a. | No |
| #33 | F | 2.1 | V | 5 | 30 | 1 | 3 | 2 | 5 | yes | I | n.a. | No |
| #34 | F | 2.1 | V | 4 | 20 | 1 | 3 | 2 | 13 | yes | I | n.a. | No |
| #35 | M | 2.1 | V | 6 | 135 | 3 | 5 | 5 | 2 | yes | II | n.a. | No |
| #36 | F | 2.2 | V | 3 | 5 | 2 | 2 | 2 | 10 | yes | I | n.a. | No |
| #37 | M | 2.2 | V | 6.5 | 90 | 2 | 4 | 2 | 8 | yes | II | n.a. | No |
| #38 | F | 2.3 | V | 7 | 55 | 2 | 2 | 1 | 2 | yes | II | n.a. | No |
| #39 | M | 2.3 | V | 4.5 | 10.7 | 3 | 6 | 6 | 18 | yes | I | n.a. | No |
| #40 | F | 2.6 | V | 3.8 | 5 | 1 | 5 | 5 | 13 | yes | I | 6.1 | No |
| #41 | F | 2.6 | V | 5.2 | 55 | 4 | 7 | 7 | 2 | yes | II | n.a. | No |
| #42 | F | 2.6 | V | 4.5 | 10 | 1 | 1 | 2 | 1 | yes | I | 6.3 | No |
| #43 | F | 2.6 | V | 2.5 | 5 | 2 | 4 | 5 | 10 | no | I | n.a. | No |
| #44 | M | 2.7 | V | 2.5 | 3 | 3 | 5 | 5 | 40 | yes | I | 4 | No |
| #45 | M | 2.7 | V | 2.5 | 3 | 3 | 5 | 5 | 40 | yes | I | 4 | No |
| #46 | F | 2.8 | V | 5.5 | 40 | 3 | 6 | 6 | 10 | yes | II | n.a. | No |
| #47 | F | 2.9 | V | 1.8 | 4 | 1 | 3 | 1 | 3 | yes | I | 5.4 | No |
| #48 | F | 3 | V + C | 6.5 | 48.5 | 1 | 5 | 5 | 13 | yes | II | 0.49 | No |
| #49 | F | 3 | V | 3 | n.a. | 1 | 3 | 1 | n.a. | no | I | n.a. | Yes |
| #50 | F | 3 | V + C | 4.5 | 20 | 3 | 3 | 4 | n.a. | yes | I | n.a. | No |
| #51 | M | 3 | V | 5.5 | 8.2 | 1 | 4 | 1 | 10 | yes | I | 0.24 | No |
| #52 | F | 3 | V | 2.5 | 7.3 | 1 | 7 | 5 | 10 | yes | I | 2.1 | No |
| #53 | F | 3 | V + C | 6 | 37.9 | 4 | 5 | 3 | 40 | yes | II | 31.8 | No |
| #54 | M | 3 | V + C | 5.5 | 30 | 2 | 4 | 4 | 40 | no | I | n.a. | No |
| #55 | M | 3 | V | 11 | 381 | 8 | 9 | 7 | n.a. | n.a. | IV | n.a. | Yes |
| #56 | F | 3 | V + C | 6 | 70 | n.a. | n.a. | n.a. | n.a. | n.a. | IV | n.a. | No |
| #57 | M | 3 | V + C | 12 | 970 | 6 | 8 | 7 | 40 | yes | III | 7.47 | No |
| #58 | F | 3.11 | V + C | 13.5 | 650 | 8 | 7 | 7 | 27 | Yes | IV | n.a. | No |
| #59 | M | 3.3 | V | 6 | 70 | n.a. | 7 | n.a. | n.a. | Yes | IV | 17 | No |
| #60 | M | 3.3 | V + C | 6 | 37.9 | 4 | 5 | 3 | 40 | Yes | III | 17 | No |
| #61 | F | 3.4 | V | 1.5 | 3.2 | 1 | 3 | 2 | 5 | Yes | I | 0.7 | No |
| #62 | F | 3.8 | V | 1.2 | 3 | 1 | 1 | 1 | n.a. | no | I | n.a. | No |
| #63 | M | 4 | V + C | 6 | 70 | 5 | 7 | 5 | 57 | Yes | II | n.a. | No |
| #64 | M | 4 | V + C | 6 | 180 | 2 | 4 | 3 | n.a. | n.a. | IV | n.a. | Yes |
| #65 | F | 4.6 | V + C | 6.5 | 70 | 3 | 5 | 4 | 57 | No | II | n.a. | Yes |
| #66 | F | 4.6 | V + C | 6.5 | 72 | 3 | 5 | 4 | 52 | Yes | III | n.a. | No |
| #67 | M | 5 | V + F + C | 4.5 | 34.4 | 6 | 3 | 2 | n.a. | Yes | I | 12.7 | No |
| #68 | F | 6 | V | 7.5 | 35 | 3 | 5 | 3 | 10 | No | II | n.a. | No |
| #69 | F | 6 | V | 4 | n.a. | 2 | 6 | 6 | 25 | Yes | I | n.a. | No |
| #70 | F | 6 | n.a. | 1 | 5 | n.a. | n.a. | n.a. | n.a. | No | II | n.a. | No |
| #71 | F | 6.2 | V + C | 7 | 55 | 5 | 8 | 6 | 11 | No | IV | n.a. | Yes |
| #72 | F | 7 | V + C | n.a. | n.a. | 0 | 0 | 0 | n.a. | No | n.a. | n.a. | Yes |
| #73 | F | 7 | V + C | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | No |
| #74 | F | 9.6 | C | 6 | 220 | 5 | 7 | 7 | 26 | No | IV | n.a. | Yes |
| #75 | M | 9.6 | V + C | 4.5 | 20 | 0 | 2 | 1 | 1 | No | I | n.a. | No |
| #76 | F | 10 | V + C | 5 | 20 | 3 | 6 | 6 | n.a. | No | II | n.a. | No |
| #77 | M | 11.5 | V + C | 2.5 | 45.6 | 0 | 2 | 2 | n.a. | Yes | I | 16.8 | No |
| #78 | F | 13 | V | 6.5 | 51 | 0 | 0 | 0 | n.a. | No | II | n.a. | No |
| #79 | M | 14 | V + C | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | No |
| #80 | F | 14.5 | C | 2 | n.a. | 0 | 1 | 0 | n.a. | No | I | n.a. | No |
| #81 | F | 15 | C | 3.2 | 20 | 0 | 2 | 1 | n.a. | No | III | n.a. | No |
| #82 | F | 15.7 | C | 7 | 60 | 1 | 2 | 3 | n.a. | No | II | n.a. | No |
| #83 | F | 16 | V + C | 20 | 1475 | 8 | 7 | 6 | n.a. | Yes | IV | 24 | Yes |
| #84 | F | 16.11 | V + C | 4 | 5 | 0 | 2 | 1 | 3 | No | IV | n.a. | No |
| #85 | M | 17 | V | 9 | 165 | 5 | 7 | 3 | n.a. | No | II | 37 | No |
| #86 | M | 17 | V | 5 | 29 | 2 | 5 | 5 | 7 | No | I | n.a. | No |
| #87 | F | 17 | V | 20 | 825 | 8 | 8 | 5 | n.a. | No | III | n.a. | Yes |
| #88 | F | 17.3 | V | 24 | 725 | 7 | 8 | 7 | n.a. | No | III | 2.7 | Yes |
| #89 | F | 17.4 | C | 10 | 5 | 5 | 8 | 7 | 40 | Yes | III | 711 | Yes |
| #90 | F | 18 | V | 13 | 1000 | 8 | 8 | 7 | n.a. | Yes | IV | n.a. | Yes |
| #91 | F | 18 | V | 12 | 280 | 6 | 8 | 7 | n.a. | n.a. | IV | n.a. | Yes |
| #92 | F | 18 | C | 2.6 | 15 | 2 | 4 | 3 | n.a. | n.a. | I | n.a. | No |
| #93 | F | 18 | n.a. | 16.5 | n.a. | n.a. | 6 | 4 | n.a. | n.a. | IV | n.a. | No |
| #94 | F | 18 | V + C | 12.5 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | IV | n.a. | Yes |
| #95 | F | 18 | C | 2.6 | 15 | 2 | 4 | 3 | n.a. | Yes | I | n.a. | No |
| Group | Weiss Score ≥ 5 | M. Weiss Score ≥ 5 | Wieneke Index ≥ 3 | MacFarlane Stage ≥ II | TNM Stage ≥ II |
|---|---|---|---|---|---|
| 1 | 35.5% | 24.2% | 17.7% | 21% | 14.5% |
| 2 | 50% | 28.6% | 28.6% | 28.6% | 28.6% |
| 3 | 55% | 50% | 55% | 40% | 40% |
| p = 0.002 | p = 0.018 | p < 0.001 | p < 0.001 | p < 0.001 | |
| HR: 6.2; CI 95% 1.2–8.3 | HR: 6.2; CI 95% 1.2–8.3 | HR: 25.8; CI 95% 1.4–3.4 | HR: 5.1; CI 95% 1–25.9 | HR: 5.1; CI 95% 1–25.9 |
| Stage | TNM | MacFarlane | MacFarlane/Sullivan |
|---|---|---|---|
| I | T1, N0, M0 | T1, N0, M0 | T1, N0, M0 |
| II | T2, N0, M0 | T2, N0, M0 | T2, N0, M0 |
| III | T1-2, N1, M0; T3, N0, M0 | T3, N0, M0 T1-3, N1, M0 (mobile nodes) | T3, N0, M0 T1-3, N1, M0 |
| IV | T3, N1, M0; T4, N0-1, M0; T1-4, N0-1, M1 | Any T, N1 or M1 (fixed nodes) | T4, N1, M0 or any M1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachega, F.S.; Suartz, C.V.; Almeida, M.Q.; Brondani, V.B.; Charchar, H.L.S.; Lacombe, A.M.F.; Martins-Filho, S.N.; Soares, I.C.; Zerbini, M.C.N.; Dénes, F.T.; et al. Retrospective Analysis of Prognostic Factors in Pediatric Patients with Adrenocortical Tumor from Unique Tertiary Center with Long-Term Follow-Up. J. Clin. Med. 2022, 11, 6641. https://doi.org/10.3390/jcm11226641
Bachega FS, Suartz CV, Almeida MQ, Brondani VB, Charchar HLS, Lacombe AMF, Martins-Filho SN, Soares IC, Zerbini MCN, Dénes FT, et al. Retrospective Analysis of Prognostic Factors in Pediatric Patients with Adrenocortical Tumor from Unique Tertiary Center with Long-Term Follow-Up. Journal of Clinical Medicine. 2022; 11(22):6641. https://doi.org/10.3390/jcm11226641
Chicago/Turabian StyleBachega, Fernanda S., Caio V. Suartz, Madson Q. Almeida, Vania B. Brondani, Helaine L. S. Charchar, Amanda M. F. Lacombe, Sebastião N. Martins-Filho, Iberê C. Soares, Maria Claudia N. Zerbini, Francisco T. Dénes, and et al. 2022. "Retrospective Analysis of Prognostic Factors in Pediatric Patients with Adrenocortical Tumor from Unique Tertiary Center with Long-Term Follow-Up" Journal of Clinical Medicine 11, no. 22: 6641. https://doi.org/10.3390/jcm11226641