
Hemostatic Abnormalities in Gaucher Disease: Mechanisms and Clinical Implications

Abstract
:1. Introduction
2. GD and Inflammation
3. Inflammation and Hemostasis
4. Glycolipids and Hemostasis
5. GD and Bleeding
5.1. Thrombocytopenia
5.2. Platelet Function Defects
5.3. Coagulopathy
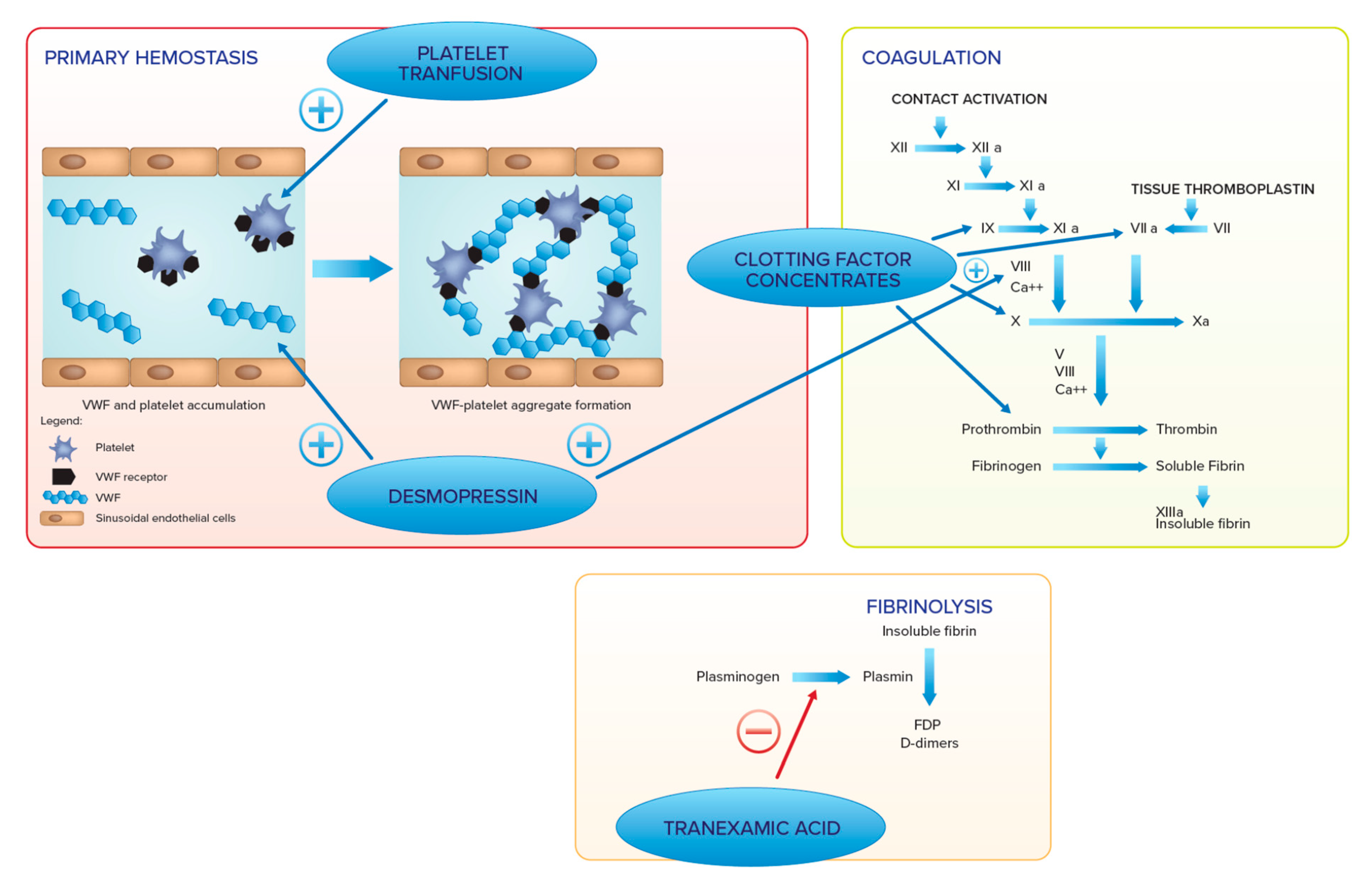
6. Hemostatic Management
7. Hemostatic Agents
8. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| β-GCase | β-glucocerebrosidase |
| BSS | Bernard–Soulier syndrome |
| Desmopressin (DDAVP) | 1-deamino-8-D-arginin-vasopressin |
| ERT | enzyme replacement therapy |
| GD | Gaucher disease |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| IL-10 | interleukin-10 |
| PF4 | platelet factor 4 |
| sIL-2R | soluble interleukin-2 receptor |
| SRT | substrate reduction therapy |
| TEG | thromboelastography |
| TNF-α | tumor necrosis factor-α |
| TNFR1 | tumor necrosis factor receptor 1 |
| TNFR2 | tumor necrosis factor receptor 2 |
References
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, G.A.; Petsko, G.A.; Kolodny, E.H. Gaucher disease. In The Online Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Cox, T.M. Gaucher disease: Understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 2001, 24 (Suppl. 2), 106–121. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. The Gaucher Registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 2000, 160, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Wenstrup, R.J.; Roca-Espiau, M.; Weinreb, N.J.; Bembi, B. Skeletal aspects of Gaucher disease: A review. Br. J. Radiol. 2002, 75 (Suppl. 1), A2–A12. [Google Scholar] [CrossRef]
- Pastores, G.M.; Barnett, N.L.; Bathan, P.; Kolodny, E.H. A neurological symptom survey of patients with type I Gaucher disease. J. Inherit. Metab. Dis. 2003, 26, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Mengel, E.; Maródi, L.; Petakov, M.; Niederau, C.; Giraldo, P.; Hughes, D.; Mrsic, M.; Mehta, A.; Hollak, C.E.M.; et al. Peripheral neuropathy in adult type 1 Gaucher disease: A 2-year prospective observational study. Brain 2010, 133, 2909–2919. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- de Fost, M.; Dahl, S.V.; Weverling, G.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E.M. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Boddupalli, C.S.; Verma, R.; Liu, J.; Yang, R.; Pastores, G.M.; Mistry, P.K.; Dhodapkar, M.V. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015, 125, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, N.J.; Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.; Wappner, R.S.; et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: A report from the Gaucher Registry. Am. J. Med. 2002, 113, 112–119. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Goldblatt, J.; Villalobos, J.; Charrow, J.; Cole, J.A.; Kerstenetzky, M.; Dahl, S.V.; Hollak, C. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab Dis 2013, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Zimran, A.; Altarescu, G.; Philips, M.; Attias, D.; Jmoudiak, M.; Deeb, M.; Wang, N.; Bhirangi, K.; Cohn, G.M.; Elstein, D. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010, 115, 4651–4656. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Brill-Almon, E.; Chertkoff, R.; Petakov, M.; Blanco-Favela, F.; Muñoz, E.T.; Solorio-Meza, S.E.; Amato, D.; Duran, G.; Giona, F.; et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011, 118, 5767–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Turkia, H.; Gonzalez, D.E.; Barton, N.W.; Zimran, A.; Kabra, M.; Lukina, E.A.; Giraldo, P.; Kisinovsky, I.; Bavdekar, A.; Ben Dridi, M.-F.; et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am. J. Hematol. 2013, 88, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Jeyakumar, M. Substrate reduction therapy. Acta Paediatr. Suppl. 2008, 97, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A. Journeys in science: Glycobiology and other paths. Annu. Rev. Biochem. 2014, 83, 1–44. [Google Scholar] [CrossRef]
- Cox, T.M.; Aerts, J.M.F.G.; Andria, G.; Beck, M.; Belmatoug, N.; Bembi, B.; Chertkoff, R.; Dahl, S.V.; Elstein, D.; Erikson, A.; et al. The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease. J. Inherit. Metab. Dis. 2003, 26, 513–526. [Google Scholar] [CrossRef] [PubMed]
- McEachern, K.A.; Fung, J.; Komarnitsky, S.; Siegel, C.S.; Chuang, W.-L.; Hutto, E.; Shayman, J.A.; Grabowski, G.A.; Aerts, J.; Cheng, S.H.; et al. A specific and potent inhibitor of glucosylceramide synthase for substrate reduction therapy of Gaucher disease. Mol. Genet. Metab. 2007, 91, 259–267. [Google Scholar] [CrossRef]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Banikazemi, M.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Rosenthal, D.I.; et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 2010, 116, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Angell, J.; Ross, L.; Puga, A.C.; et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4 years of treatment. Blood Cells Mol. Dis. 2014, 53, 274–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, R.S.; Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Aguzzi, R.; Puga, A.C.; et al. Skeletal improvement in patients with Gaucher disease type 1: A phase 2 trial of oral eliglustat. Skelet. Radiol. 2014, 43, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Amato, D.; Baris, H.; Dasouki, M.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: The ENGAGE randomized clinical trial. JAMA 2015, 313, 695–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.M.; Drelichman, G.; Cravo, R.; Balwani, M.; Burrow, T.A.; Martins, A.M.; Lukina, E.; Rosenbloom, B.; Ross, L.; Angell, J.; et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: A phase 3, randomised, open-label, non inferiority trial. Lancet 2015, 385, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.-L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Halene, S.; Yang, M.; Iqbal, J.; Yang, R.; Mehal, W.Z.; Chuang, W.L.; Jain, D.; Yuen, T.; Sun, L.; et al. Gaucher disease gene GBA functions in immune regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 10018–10023. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M. Lysosomes, lysosomal storage disease, and inflammation. J. Inborn Errors Metab. Screen. 2016, 4, 465. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar]
- Allen, M.; Myer, B.; Khokher, A.; Rushton, N.; Cox, T. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM 1997, 90, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakakis, H.; Spanou, C.; Kondyli, A.; Dimitriou, E.; Van Weely, S.; Hollak, C.E.; Van Oers, M.H.; Aerts, J. Plasma tumor-necrosis factor α (TNF-α levels in Gaucher disease. Biochim. Biophys. Acta 1996, 1317, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Hollak, C.E.; Evers, L.; Aerts, J.; van Oers, M.H. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol. Dis. 1997, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- van Breemen, M.J.; de Fost, M.; Voerman, J.S.; Laman, J.D.; Boot, R.; Maas, M.; Hollak, C.E.; Aerts, J.M.; Rezaee, F. Increased plasma macrophage inflammatory protein (MIP)-1α and MIP-1β levels in type 1 Gaucher disease. Biochim. Biophys. Acta 2007, 1772, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Hollak, C.E.; Van Weely, S.; Van Oers, M.H.; Aerts, J. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Grabowsky, G.A. Immunological cells and functions in Gaucher disease. Crit. Rev. Oncog. 2013, 18, 197–220. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; Maiolo, A. Cytokines and hemostasis. Haematologica 2000, 85, 967–972. [Google Scholar]
- Levi, M.; van der Poll, T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc. Med. 2005, 15, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Bester, J.; Pretorius, E. Effects of IL-1B, IL-6 and IL-8 on erythrocites, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188–32198. [Google Scholar] [CrossRef] [Green Version]
- Page, M.; Bester, J.; Pretorius, E. Interleukin-12 and its procoagulant effect on erythrocytes, platelets and fibrin(ogen): The lesser known side of inflammation. Br. J. Haematol. 2018, 180, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF-a and complement component 3 on coagulation. 3 on coagulation. Sci. Rep. 2018, 8, 1812–1820. [Google Scholar] [CrossRef]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jäckel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Grabowski, G.A.; Kohl, J. An unexpected player in Gaucher disease: The multiple roles of complement in disease development. Semin. Immunol. 2018, 37, 30–42. [Google Scholar] [CrossRef]
- Maouia, A.; Rebetz, J.; Kapur, R.; Semple, J.W. The immune nature of platelets revised. Transf. Med. Rev. 2020, 34, 209–220. [Google Scholar] [CrossRef]
- Deguchi, H.; Yegneswaran, S.; Griffin, J.H. Sphingolipids as bioactive regulators of thrombin generation. J. Biol. Chem. 2004, 279, 12036–12042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguchi, H.; Fernadez, J.A.; Griffin, J.H. Neutral glycosphingolipid-dependent inactivation of coagulation factor Va by activated protein C and protein S. J. Biol. Chem. 2002, 277, 8861–8865. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, H.; Bouma, B.N.; Middeldorp, S.; Lee, Y.M.; Griffin, J.H. Decreased plasma sensitivity to activated protein C by oral contraceptives is associated with decreases in plasma glucosylceramide. J. Thromb. Haemost. 2005, 3, 935–938. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Haematol. 2015, 90 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef]
- Givol, N.; Goldstein, G.; Peleg, O.; Shenkman, B.; Zimran, A.; Elstein, D.; Kenet, G. Thrombocytopenia and bleeding in dental procedures of patients with Gaucher disease. Haemophilia 2012, 18, 117–121. [Google Scholar] [CrossRef]
- Katz, K.; Tamary, H.; Lahav, J.; Soudry, M.; Cohen, I.J. Increased operative bleeding during orthopaedic surgery in patients with type I Gaucher disease and bone involvement. Bull. Hosp. Jt. Dis. 1999, 58, 188–190. [Google Scholar]
- Mitrovic, M.; Elezovic, I.; Grujicic, D.; Miljic, P.; Suvajdzic, N. Complex haemostatic abnormalities as a cause of bleeding after neurosurgery in a patient with Gaucher disease. Platelets 2015, 26, 260–262. [Google Scholar] [CrossRef]
- Granovsky-Grisaru, S.; Belmatoug, N.; Dahl, S.V.; Mengel, E.; Morris, E.; Zimran, A. The management of pregnancy in Gaucher disease. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 156, 3–8. [Google Scholar] [CrossRef]
- Zimran, A.; Morris, E.; Mengel, E.; Kaplan, P.; Belmatoug, N.; Hughes, D.A.; Malinova, V.; Heitner, R.; Sobreira, E.; Mrsić, M.; et al. The female Gaucher patients: The impact of enzyme replacement therapy around key reproductive events (menstruation, pregnancy and menopause. Blood Cells Mol. Dis. 2009, 43, 264–288. [Google Scholar] [CrossRef]
- Selton, J.; Perrin, J.; Ropion, H.; Abdelfatah, M.; Siouala, B.; Pruna, L.; Kaminsky, P. Iliopsoas Hematoma in Gaucher’s Disease. Intern. Med. 2011, 50, 2643–2647. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, P.; Andersson, H.C.; Kacena, K.A.; Yee, J.D. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch. Pediatr. Adolesc. Med. 2006, 160, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, G.A.; Barton, N.W.; Pastores, G.; Dambrosia, J.M.; Banerjee, T.K.; McKee, M.A.; Parker, C.; Schiffmann, R.; Hill, S.C.; Brady, R.O. Enzyme therapy in type 1 Gaucher disease: Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann. Intern. Med. 1995, 122, 33–39. [Google Scholar] [CrossRef]
- Stein, P.; Malhotra, A.; Haims, A.; Pastores, G.M.; Mistry, P.K. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage-targeted enzyme replacement therapy. J. Inherit. Metab. Dis. 2010, 33, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Hollak, C.E.M.; Belmatoug, N.; Cole, J.A.; Dahl, S.V.; Deegan, P.B.; Goldblatt, J.; Rosenbloom, B.; Dussen, L.; Tylki-Szymańska, A.; Weinreb, N.J.; et al. Characteristics of type I Gaucher disease associated with persistent thrombocytopenia after treatment with imiglucerase for 4-5 years. Br. J. Haematol. 2012, 158, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Linari, S.; Castaman, G. Hematological manifestations and complications of Gaucher disease. Expert Rev. Hematol. 2016, 9, 51–58. [Google Scholar] [CrossRef]
- Rosenbaum, H. Hemorragic aspects of Gaucher disease. Rambam Maimonides Med. J. 2014, 5, e003932. [Google Scholar] [CrossRef] [Green Version]
- Spectre, G.; Roth, B.; Ronen, G.; Rosengarten, D.; Elstein, D.; Zimran, A.; Varon, D.; Revel-Vilk, S. Platelet adhesion defect in type 1 Gaucher disease is associated with risk of mucosal bleeding. Br. J. Haematol. 2011, 153, 372–378. [Google Scholar] [CrossRef]
- Nilsson, O.; Håkansson, G.; Dreborg, S.; Groth, C.G.; Svennerhoim, L. Increased cerebroside concentration in plasma erythrocytes in Gaucher disease: Significant differences between type I and type III. Clin. Genet. 1982, 22, 274–279. [Google Scholar] [CrossRef]
- Aerts, J.M.; Hollak, C.E. Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clin. Haematol. 1997, 10, 691–709. [Google Scholar] [CrossRef]
- Gousset, K.; Wolkers, W.F.; Tsvetkova, N.M.; Oliver, A.E.; Field, C.L.; Walker, N.J.; Crowe, J.H.; Tablin, F. Evidence for a physiological role for membrane rafts in human platelets. J. Cell. Physiol. 2002, 190, 117–128. [Google Scholar] [CrossRef]
- Kelsey, H.; Christopoulos, C.; Gray, A.A.; Machin, S.J. Acquired pseudo-pseudo Bernard-Soulier syndrome complicating Gaucher’s disease. J. Clin. Pathol. 1994, 47, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Berndt, M.C.; Andrews, R.K. Bernard-Soulier syndrome. Haematologica 2011, 96, 355–359. [Google Scholar] [CrossRef]
- Giona, F.; Palumbo, G.; Amendola, A.; Santoro, C.; Mazzuconi, M.G. Platelet Function Abnormalities in Gaucher Disease Patients. Am. J. Hematol. 1999, 61, 103–106. [Google Scholar] [CrossRef]
- Giona, F.; Palumbo, G.; Amendola, A.; Santoro, C.; Mazzuconi, M.G. Platelet function and coagulation abnormalities in type 1 Gaucher disease patients: Effects of enzyme replacement therapy (ERT). J. Thromb. Haemost. 2006, 4, 1831–1833. [Google Scholar] [CrossRef]
- Mitrovic, M.; Antic, D.; Elezovic, I.; Janic, D.; Miljic, P.; Sumarac, Z.; Nikolic, T.; Suvajdzic, N. Haemostatic abnormalities in treatment-naïve patients with type 1 Gaucher’s disease. Platelets 2012, 23, 143–149. [Google Scholar] [CrossRef]
- Komninaka, V.; Repa, K.; Marinakis, T.; Pouliakis, A.; Koutsouri, T.; Tsokanas, D.; Flevary, P.; Voskaridou, E.; Politou, M. Platelet function defects in patients with Gaucher disease on long term ERT implications for evaluation at bleeding challenges. Blood Cells Mol. Dis. 2020, 80, 102371. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Naamad, M.; Frydman, D.; Freund, M.R.; Dinur, T.; Istaiti, M.; Becker-Cohen, M.; Falk, R.; Broide, E.; Michelson, A.D.; et al. Platelet Activation and Reactivity in a Large Cohort of Patients with Gaucher Disease. Thromb. Haemost. 2022, 122, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; Levi, M.; Berends, F.; Aerts, J.M.F.G.; van Oers, M.H.J. Coagulation. abnormalities in type 1 Gaucher disease are due to low-grade activation and can be partly restored by enzyme supplementation therapy. Br. J. Haematol. 1997, 96, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Boklan, B.F.; Sawitsky, A. Factor IX deficiency in Gaucher disease. An in vitro phenomenon. Arch. Intern. Med. 1976, 136, 489–492. [Google Scholar] [CrossRef]
- Billett, H.H.; Rizvi, S.; Sawitsky, A. Coagulation abnormalities in patients with Gaucher’s Disease: Effect of therapy. Am. J. Hematol. 1996, 51, 234–236. [Google Scholar] [CrossRef]
- Barone, R.; Giuffrida, G.; Musso, R.; Carpinteri, G.; Fiumara, A. Haemostatic abnormalities and lupus anticoagulant activity in patients with Gaucher disease type I. Inherit. Metab. Dis. 2000, 23, 387–390. [Google Scholar] [CrossRef]
- Deghady, A.; Marzouk, I.; El-Shayeb, A.; Wali, Y. Coagulation abnormalities in type 1 Gaucher disease in children. Pediatr. Hematol. Oncol. 2006, 23, 411–417. [Google Scholar] [CrossRef]
- Serratrice, C.; Cherin, P.; Lidove, O.; Noel, E.; Masseau, A.; Leguy-Seguin, V.; Jaussaud, R.; Marie, I.; Lavigne, C.; Maillot, F. Coagulation Parameters in Adult Patients with Type-1 Gaucher Disease. J. Hematol. 2019, 8, 121–124. [Google Scholar] [CrossRef]
- Mitrovic, M.; Elezovic, I.; Miljic, P.; Suvajdzic, N. Acquired von Willebrand syndrome in patients with Gaucher disease. Blood Cells Mol. Dis. 2014, 52, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Berrebi, A.; Malnick, S.D.; Vorst, E.J.; Stein, D. High incidence of factor XI deficiency in Gaucher’s disease. Am. J. Haematol. 1992, 40, 153. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.; Puliafito, C.A. Peripheral retinal vascular lesions in a patient with Gaucher disease and factor XI deficiency. Arch. Ophthalmol. 1988, 106, 1351–1352. [Google Scholar] [CrossRef]
- Benjamin, D.; Joshua, H.; Douer, D.; Shaklai, M.; Krugliac, Y.; Pinkhas, J. Circulating anticoagulant in patients with Gaucher’s disease. Acta Haematol. 1979, 61, 223–226. [Google Scholar] [CrossRef]
- Favaloro, E.J. Clinical utility of the PFA-100. Semin. Thromb. Hemost. 2008, 34, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Ioschovich, A.; Fadeev, D.; Kenet, G.; Naamad, M.; Schtrechman, G.; Zimran, A.; Elstein, D. Thromboelastography as a surrogate marker of perisurgical hemostasis in Gaucher Disease. Clin. Appl. Thromb. Hemost. 2016, 22, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Nogami, K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br. J. Haematol. 2016, 174, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitlur, M.; Sorensen, B.; Rivard, G.E.; Young, G.; Ingerslev, J.; Othman, M.; Nugent, D.; Kenet, G.; Escobar, M.; Lusher, J. Standardization of thromboelastography: A report from the TEG-ROTEM working group. Haemophilia 2011, 17, 532–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengborn, L.; Blomback, M. Tranexamic acid—An old drug still going strong and making a revival. Thromb. Res. 2015, 135, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.D.; Gueye, N.A. The medical management of abnormal uterine bleeding in reproductive-aged women. Am. J. Obstet. Gynecol. 2016, 214, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Huq, F.Y. Levonorgestrel-releasing intrauterine system for the management of heavy menstrual bleeding in women with inherited bleeding disorders: Long-term follow-up. Contraception 2011, 83, 242–247. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Ruggeri, Z.M.; Pareti, F.I.; Capitanio, A. 1-Deamino-8-d-arginine vasopressin: A new pharmacological approach to the management of haemophilia and von Willebrands’ diseases. Lancet 1977, 1, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Kentro, T.B.; Lottenberg, R.; Kitchens, C.S. Clinical efficacy of desmopressin acetate for hemostatic control in patients with primary platelet disorders undergoing surgery. Am. J. Hematol. 1987, 24, 215–219. [Google Scholar] [CrossRef] [PubMed]
- DiMichele, D.M.; Hathaway, W.E. Use of DDAVP in inherited and acquired platelet dysfunction. Am. J. Hematol. 1990, 33, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Ghosh, S.; Sun, L.; Yang, X.; Disa, J.; Pickens, P.; Polansky, M. Mechanisms of platelet dysfunction and response to DDAVP in patients with congenital platelet function defects. A double-blind placebo-controlled trial. Thromb. Haemost. 1995, 74, 1071–1078. [Google Scholar]
- Fuse, I.; Higuchi, W.; Mito, M.; Aizawa, Y. DDAVP normalized the bleeding time in patients with congenital platelet TxA2 receptor abnormality. Transfusion 2003, 43, 563–567. [Google Scholar] [CrossRef]
- Coppola, A.; Di Minno, G. Desmopressin in inherited disorders of platelet function. Haemophilia 2008, 14 (Suppl. 1), 31–39. [Google Scholar] [CrossRef]
- Tosetto, A.; Balduini, C.; Cattaneo, M.; De Candia, E.; Mariani, G.; Molinari, A.; Rossi, E.; Siragusa, S. Italian Society for Haemostasis and Thrombosis. Management of bleeding and of invasive procedures in patients with platelet disorders and/or thrombocytopenia: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb. Res. 2009, 124, e13–e18. [Google Scholar] [CrossRef]
- Cattaneo, M. Desmopressin in the treatment of patients with defects of platelet function. Haematologica 2002, 87, 1122–1124. [Google Scholar] [PubMed]
- Parker, R.I.; Grewal, R.P.; McKeown, L.P.; Barton, N.W. Effect of platelet count on the DDAVP-induced shortening of bleeding time in thrombocytopenic Gaucher’s patients. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 39–43. [Google Scholar] [CrossRef]
- Colucci, G.; Stutz, M.; Rochat, S.; Conte, T.; Pavicic, M.; Reusser, M.; Giabbani, E.; Huynh, A.; Thürlemann, C.; Keller, P.; et al. The effect of desmopressin on platelet function: A selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood 2014, 123, 1905–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannucci, P.M.; Lusher, J.M. Desmopressin and thrombosis. Lancet 1989, 2, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Cappellini, M.D.; Berger, M.; van Droogenbroeck, J.; de Fost, M.; Janic, D.; Marinakis, T.; Rosenbaum, H.; Villarubia, J.; Zhukovskaya, E.; et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br. J. Haematol. 2007, 138, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elstein, D.; Renbaum, P.; Levy-Lahad, E.; Zimran, A. Incidence of thrombophilia in patients with Gaucher disease. Am. J. Med. Genet. 2000, 95, 429–431. [Google Scholar] [CrossRef]
- Shitrit, D.; Rudensky, B.; Zimran, A.; Elstein, D. D-dimer assay in Gaucher disease: Correlation with severity of bone and lung involvement. Am. J. Hematol. 2003, 73, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Sherif, E.M.; Tantawy, A.A.; Adly, A.A.; Kader, H.A.; Ismail, E.A. D-dimer assay in Egyptian patients with Gaucher disease: Correlation with bone and lung involvement. Blood Coagul. Fibrinolysis 2011, 22, 176–184. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Subtype | Incidence | Ethnic Group | Age at Onset | Primary CNS Involvement | Splenomegaly and/or Hepatomegaly | Cytopenia 1 | Bone Disease 2 | Other |
|---|---|---|---|---|---|---|---|---|
| Type 1 | 1:40,000–1:60,000 1:450 in Ashkenazi Jews | Panethnic, more common in Ashkenazi Jews | Any age | No | Yes | Yes | Yes | Pulmonary disease |
| Type 2 (Acute or infantile) | <1:100,000 | Panethnic | Infancy—early childhood | Bulbar signs Pyramidal signs Cognitive impairment | Yes | Yes | No | Pulmonary disease Dermatologic changes |
| Type 3 (Subacute, juvenile) | <1:50,000 to <1:100,000 | Panethnic | Childhood | Oculomotor apraxia Seizures Progressive myoclonic epilepsy | Yes | Yes | Yes | Pulmonary disease |
| Perinatal lethal form | <1:10,000 | Panethnic | Perinatal | Pyramidal signs | No | No | No | Ichthyosiform or collodion skin changes Nonimmune hydrops fetalis |
| Reference | Gillis et al., 1999 [70] | Giona et al., 2006 [71] | Spectre et al., 2011 [64] | Mitrovic et al., 2012 [72] | Komninaka et al., 2020 [73] | Revel-Vilk et al., 2021 [74] |
|---|---|---|---|---|---|---|
| Patients (N) | 32 | 13 | 48 | 31 | 29 | 149 |
| Bleeding manifestations | 12.5% | 15% | 58% | 32% | 79% | 49% |
| Plts (×109 L−1) (median; range) | 180 (74–508) | 142 (51–284) | 193 (153–252) | 108 | 147 (103–192) | 176 (30–485) |
| Plt adhesion deficiency | - | - | 66% | - | - | - |
| Plt aggregation abnormalities | 22% | 46% | 22% | 61% | - | - |
| PFA Collagen/EPI prolonged PFA Collagen/ADP prolonged PFA Collagen/EPI + Collagen/ADP prolonged | - - - | - - - | - - - | - - - | 55% 79% 52% | - - - |
| Platelet reactivity deficiency | - | - | - | - | - | 53% |
| Reference | Boklan et al. (1976) [76] | Billett et al. (1996) [77] | Hollak et al. (1997) [75] | Katz et al. (1999) [53] | Barone et al. (2000) [78] | Giona et al. (2006) [71] | Deghady et al. (2006) [79] | Mitrovic et al. (2012) [72] | Serratrice et al. (2019) [80] | Komninaka et al. (2020) [73] |
|---|---|---|---|---|---|---|---|---|---|---|
| Patients (N) | 11 | 9 | 30 | 28 | 5 | 15 | 10 | 31 | 43 | 29 |
| ↑ PT | - | 11% | 42% | 81% | 80% | 47% | 100% | 52% | 12% | 0% |
| ↑ APTT | 100% | 55% | 38% | - | 100% | 53% | 60% | 42% | 21% | 7% |
| ↑ Fibrinogen | - | 0 | - | - | - | - | - | - | 0 | 7% |
| ↓ FII | - | 11% | 50% | - | 20% | - | 20% | - | 8% | - |
| ↓ FV | 18% | 22% | 87% | 27% | 100% | 23% | 30% | - | 31% | - |
| ↓ FVII | 9% | - | 33% | - | 0 | 8% | 50% | - | 3.4% | - |
| ↓ FVIII | 27% | 11% | 10% | 27% | 60% | 15% | 30% | - | 4% | 0 |
| ↓ FIX | 73% | 0 | 3% | 14% | 20% | 31% | 20% | - | 4.50 | - |
| ↓ FX | 9% | - | 57% | - | 20% | 8% | 10% | - | 11.50 | - |
| ↓ FXI | - | 33% | 27% | 36% | 40% | 0 | 0 | - | 20% | - |
| ↓ FXII | - | - | 30% | 27% | 25% | 15% | 10% | - | 13% | - |
| ↓ VWF | - | 11% | - | - | - | 0 | - | - | 0 | |
| ↓ ADAMST13 | - | - | - | - | - | - | - | - | 0 | 48% |
| ↓ PC | - | - | 26% | - | - | - | - | 10% | 11.5% | 0 |
| ↓ PS | - | - | 11% | - | - | - | - | - | 19% | 0 |
| ↓ AT | - | - | 3% | - | - | - | - | 0 | 0 | 0 |
| ↑ D dimer | - | - | 68% | - | - | - | - | 76% | - | 38% |
| ↑ TAT | - | - | 46% | - | - | - | - | 43% | - | - |
| ↓ PAI | - | - | - | - | - | - | - | - | - | 69% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linari, S.; Castaman, G. Hemostatic Abnormalities in Gaucher Disease: Mechanisms and Clinical Implications. J. Clin. Med. 2022, 11, 6920. https://doi.org/10.3390/jcm11236920
Linari S, Castaman G. Hemostatic Abnormalities in Gaucher Disease: Mechanisms and Clinical Implications. Journal of Clinical Medicine. 2022; 11(23):6920. https://doi.org/10.3390/jcm11236920
Chicago/Turabian StyleLinari, Silvia, and Giancarlo Castaman. 2022. "Hemostatic Abnormalities in Gaucher Disease: Mechanisms and Clinical Implications" Journal of Clinical Medicine 11, no. 23: 6920. https://doi.org/10.3390/jcm11236920