
Freshwater Clam Extract Mitigates Neuroinflammation and Amplifies Neurotrophic Activity of Glia: Insights from In Vitro Model of Neurodegenerative Pathomechanism

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Freshwater Clam Extract (FCE)
2.2. Preparation of Primary Glial Cell Cultures
2.3. In Vitro Models for Injury-Induced Neurodegeration
2.4. Cell Viability Analysis
2.5. Measurement of Intracellular ROS
2.6. Flow Cytometric Analysis of Cell Cycle Distribution
2.7. Flow Cytometry Analysis of Cell Apoptosis (Annexin V/Propidium Iodide Assay)
2.8. Nitric Oxide Assay
2.9. TNF-α Assay
2.10. Real-Time Quantitative RT-PCR Analysis (qRT-PCR) for Primary Glial Cells
2.11. Statistical Analysis
3. Results
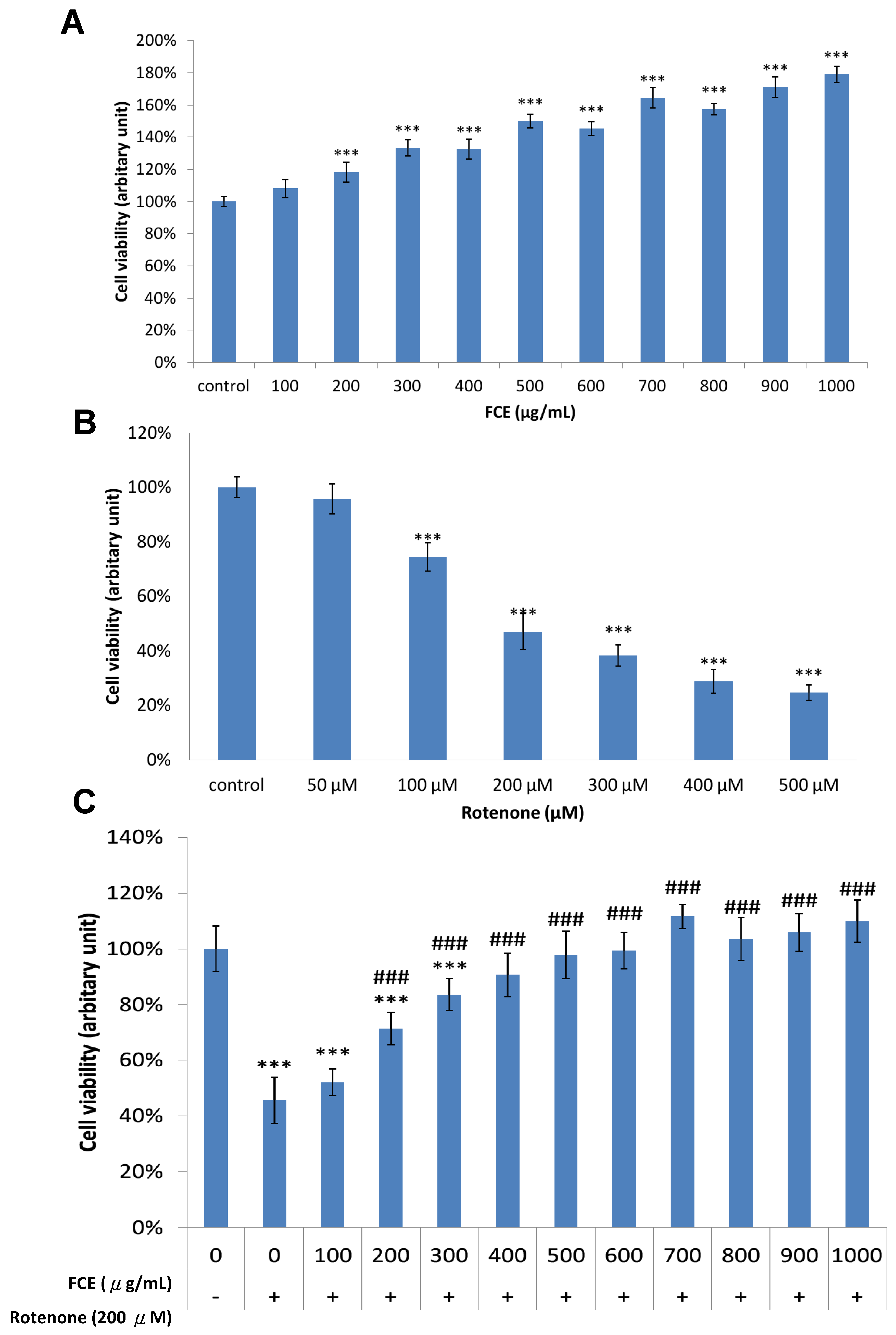
3.1. Pharmacological Effect of FCE and Rotenone on Cell Survival in Primary Glial Cells
3.2. FCE Protected against Rotenone-Induced Cytotoxicity in Primary Glial Cells
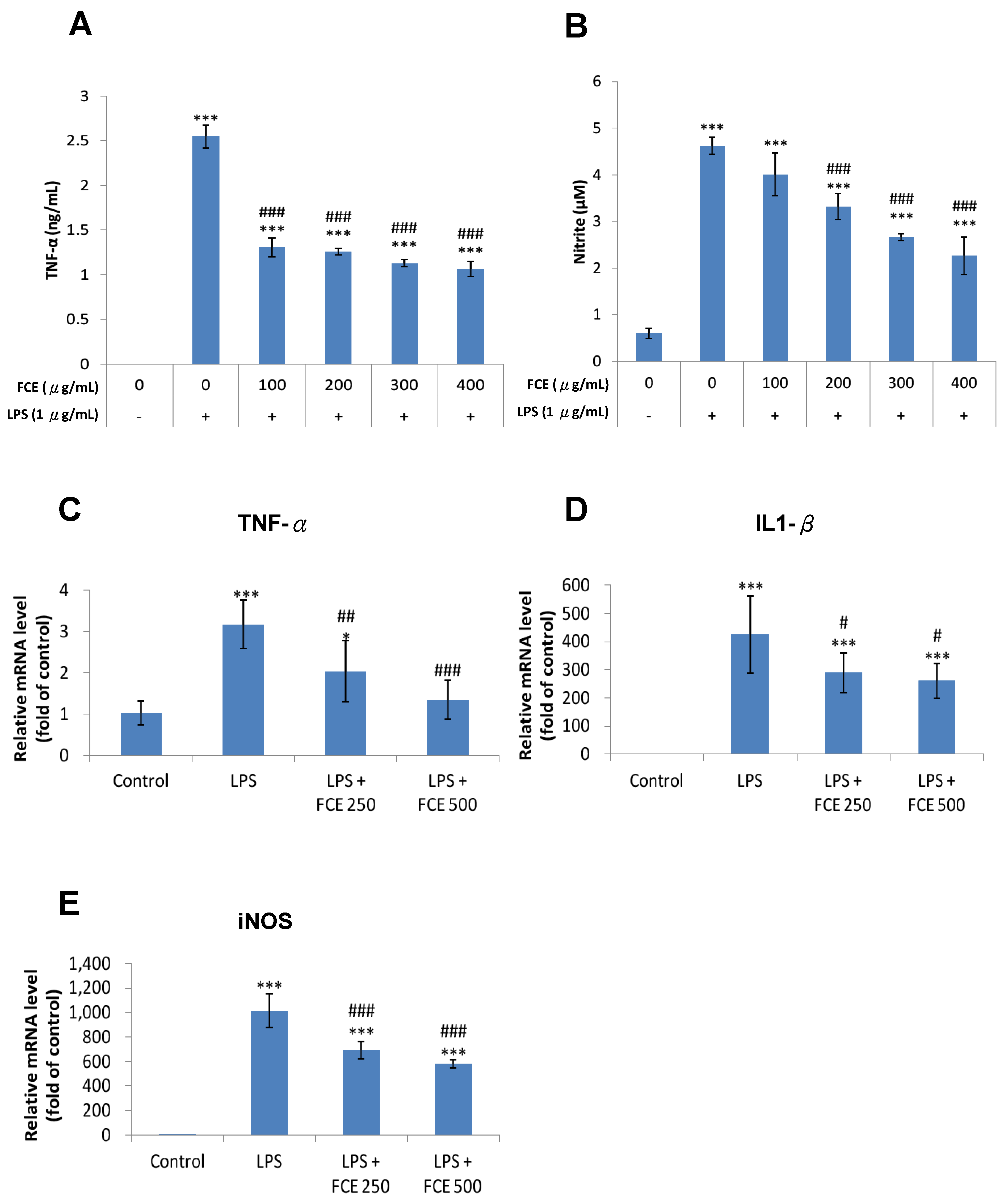
3.3. FCE Exerted Promising Anti-Inflammatory Effects against Injury-Induced Neuroinflammation in Primary Glial Cells
3.4. FCE Diminished Pro-Inflammatory mRNA Expression of TNF-α, iNOS and IL-1 Triggered by LPS in Primary Glial Cells
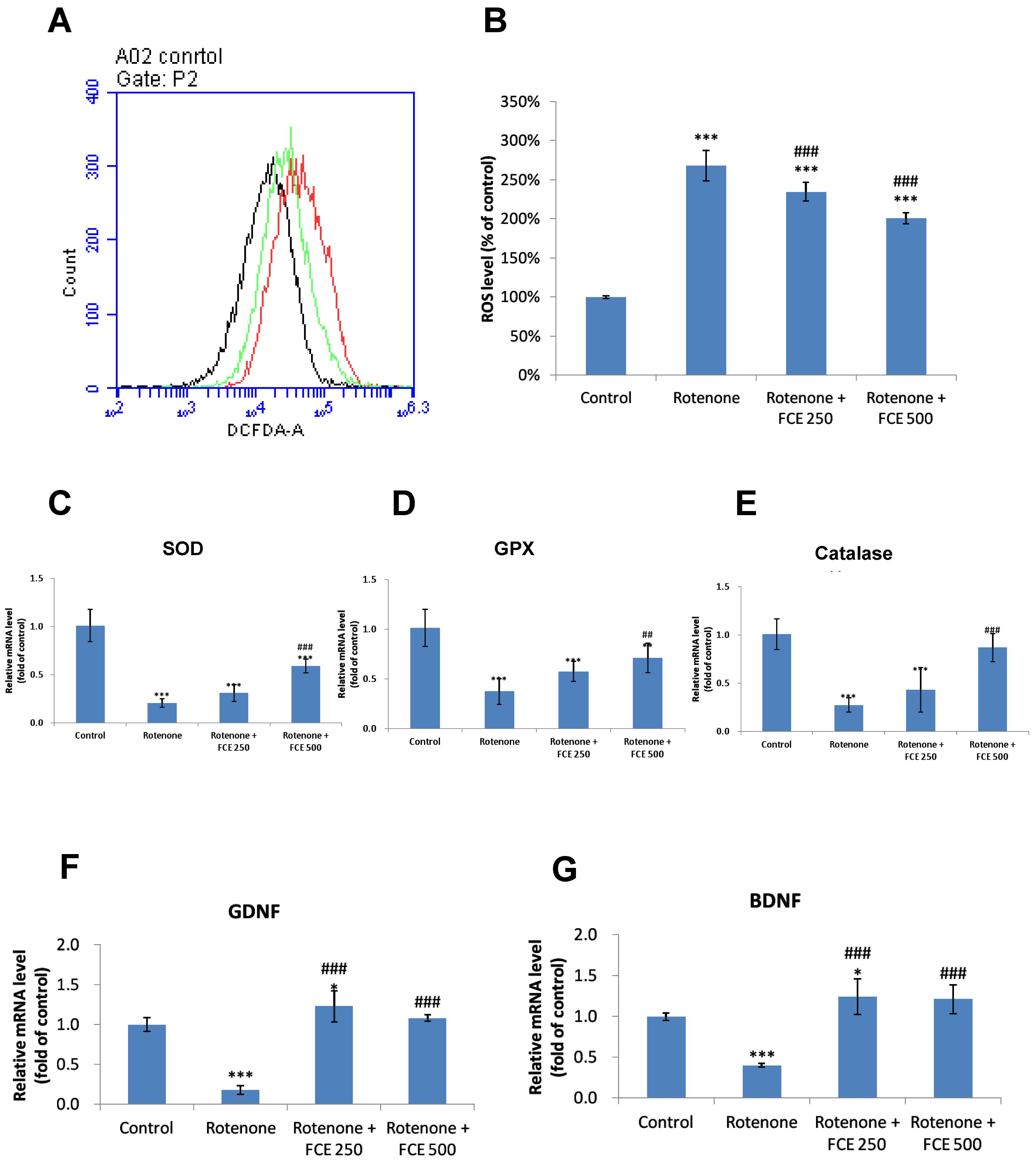
3.5. FCE Attenuated Rotenone-Induced Oxidative Stress in Primary Glial Cells
3.6. FCE Halted Rotenone-Induced Decrease in mRNA Expression of Antioxidant Enzymes in Primary Glial Cells
3.7. FCE Augmented mRNA Expression of Neurotrophic Factors in Primary Glial Cells
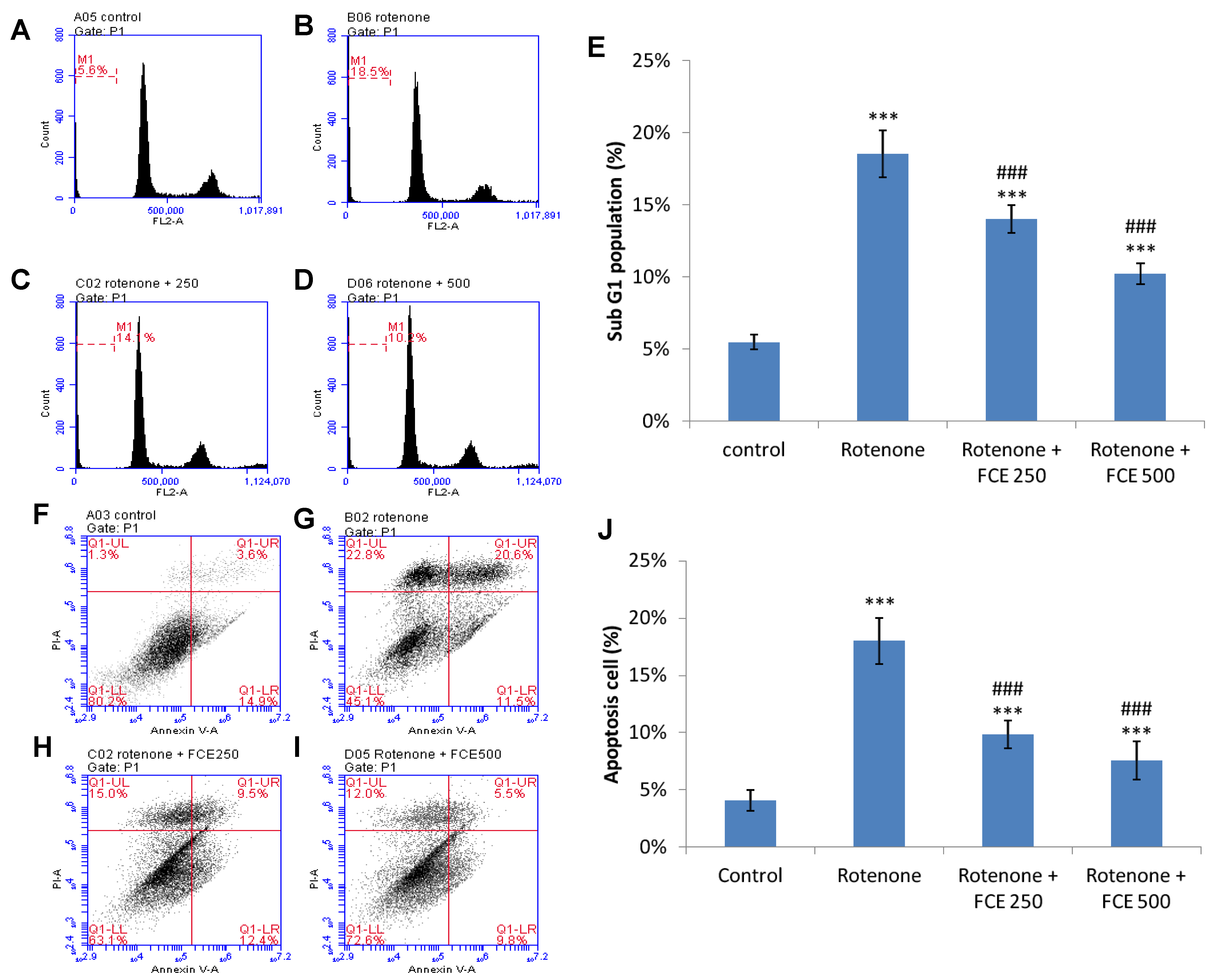
3.8. FCE Protected against Rotenone-Induced Apoptosis in Primary Glial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Vartiainen, N.E.; Ying, W.; Chan, P.H.; Koistinaho, J.; Swanson, R.A. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 2001, 77, 1601–1610. [Google Scholar] [CrossRef]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escartin, C.; Valette, J.; Lebon, V.; Bonvento, G. Neuron-astrocyte interactions in the regulation of brain energy metabolism: A focus on NMR spectroscopy. J. Neurochem. 2006, 99, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorklund, A.; Dunnett, S.B.; Brundin, P.; Stoessl, A.J.; Freed, C.R.; Breeze, R.E.; Levivier, M.; Peschanski, M.; Studer, L.; Barker, R. Neural transplantation for the treatment of Parkinson’s disease. Lancet Neurol. 2003, 2, 437–445. [Google Scholar] [CrossRef]
- Choi-Lundberg, D.L.; Lin, Q.; Chang, Y.N.; Chiang, Y.L.; Hay, C.M.; Mohajeri, H.; Davidson, B.L.; Bohn, M.C. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 1997, 275, 838–841. [Google Scholar] [CrossRef]
- Niranjan, R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: Focus on astrocytes. Mol. Neurobiol. 2014, 49, 28–38. [Google Scholar] [CrossRef]
- Emborg, M.E. Evaluation of animal models of Parkinson’s disease for neuroprotective strategies. J. Neurosci. Methods 2004, 139, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism. Relat Disord. 2012, 18, S210–S212. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.Y.; Wood, W.G. Recent developments in understanding oxidative mechanisms and contributions of glial cell activation, mitochondrial dysfunction, and lipids and signaling pathways to neurodegenerative diseases. Preface. Mol. Neurobiol. 2010, 41, 53–54. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, G.E.; Iarkov, A.; Moran, V.E. Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 340. [Google Scholar]
- Chijimatsu, T.; Umeki, M.; Okuda, Y.; Yamada, K.; Oda, H.; Mochizuki, S. The fat and protein fractions of freshwater clam ( Corbicula fluminea) extract reduce serum cholesterol and enhance bile acid biosynthesis and sterol excretion in hypercholesterolaemic rats fed a high-cholesterol diet. Br. J. Nutr. 2011, 105, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Chijimatsu, T.; Tatsuguchi, I.; Oda, H.; Mochizuki, S. A Freshwater clam (Corbicula fluminea) extract reduces cholesterol level and hepatic lipids in normal rats and xenobiotics-induced hypercholesterolemic rats. J. Agric. Food Chem. 2009, 57, 3108–3112. [Google Scholar] [CrossRef]
- Hsu, C.L.; Hsu, C.C.; Yen, G.C. Hepatoprotection by freshwater clam extract against CCl4-induced hepatic damage in rats. Am. J. Chin. Med. 2010, 38, 881–894. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Lin, M.S.; Hua, K.F.; Chen, W.J.; Lin, C.C. Neuroprotection by freshwater clam extract against the neurotoxin MPTP in C57BL/6 mice. Neurosci. Lett. 2017, 642, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.F.; Chen, G.M.; Ho, C.L.; Chen, M.C.; Chen, Y.L.; Chen, W.J.; Huang, J.F.; Perng, Y.S.; Lin, C.C. Freshwater clam extract inhibits inflammatory responses in LPS-activated macrophages by reducing the activation of mitogen-activated protein kinases and NF-kappaB. Nat. Prod. Commun. 2012, 7, 1435–1440. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.S.; Sun, Y.Y.; Chiu, W.T.; Hung, C.C.; Chang, C.Y.; Shie, F.S.; Tsai, S.H.; Lin, J.W.; Hung, K.S.; Lee, Y.H. Curcumin Attenuates the Expression and Secretion of RANTES after Spinal Cord Injury In Vivo and Lipopolysaccharide-Induced Astrocyte Reactivation In Vitro. J. Neurotrauma 2011, 28, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chiang, T.H.; Chen, W.J.; Sun, Y.Y.; Lee, Y.H.; Lin, M.S. CISD2 serves a novel role as a suppressor of nitric oxide signalling and curcumin increases CISD2 expression in spinal cord injuries. Injury 2015, 46, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–7015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miriam, M.; Paula, M.I.; Leyre, M.; Miriam, H.; Jose, B.; Carmen, G. An easy and fast way to obtain a high number of glial cells from rat cerebral tissue: A beginners approach. Protoc. Exch. 2011, 218. [Google Scholar]
- Radad, K.; Rausch, W.D.; Gille, G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 2006, 49, 379–386. [Google Scholar] [CrossRef]
- Liang, Y.; Jing, X.; Zeng, Z.; Bi, W.; Chen, Y.; Wu, X.; Yang, L.; Liu, J.; Xiao, S.; Liu, S.; et al. Rifampicin attenuates rotenone-induced inflammation via suppressing NLRP3 inflammasome activation in microglia. Brain Res. 2015, 1622, 43–50. [Google Scholar] [CrossRef]
- Miller, R.L.; James-Kracke, M.; Sun, G.Y.; Sun, A.Y. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res. 2009, 34, 55–65. [Google Scholar] [CrossRef]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garcia-Garrote, M.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. NRF2 Activation and Downstream Effects: Focus on Parkinson’s Disease and Brain Angiotensin. Antioxidants 2021, 10, 1649. [Google Scholar] [CrossRef]
- Chen, J.; Li, M.; Zhou, X.; Xie, A.; Cai, Z.; Fu, C.; Peng, Y.; Zhang, H.; Liu, L. Rotenone-Induced Neurodegeneration Is Enabled by a p38-Parkin-ROS Signaling Feedback Loop. J. Agric. Food Chem. 2021, 69, 13942–13952. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, L.; Jiao, Y.; Zhang, Y.; Wang, Y.; Zhu, K.; Sun, C. Thioredoxin-1 mediates neuroprotection of Schisanhenol against MPP(+)-induced apoptosis via suppression of ASK1-P38-NF-κB pathway in SH-SY5Y cells. Sci. Rep. 2021, 11, 21604. [Google Scholar] [CrossRef] [PubMed]
- Strycharz-Dudziak, M.; Kiełczykowska, M.; Drop, B.; Świątek, Ł.; Kliszczewska, E.; Musik, I.; Polz-Dacewicz, M. Total Antioxidant Status (TAS), Superoxide Dismutase (SOD), and Glutathione Peroxidase (GPx) in Oropharyngeal Cancer Associated with EBV Infection. Oxid. Med. Cell Longev. 2019, 2019, 5832410. [Google Scholar]
- Ekor, M. The growing use of herbal medicines: Issues relating to adverse reactions and challenges in monitoring safety. Front. Pharmacol. 2014, 4, 177. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.K.; Venkatesh, P.; Ponnusankar, S. Ethnopharmacology and integrative medicine–Let the history tell the future. J. Ayurveda. Integr. Med. 2010, 1, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.C.; Wu, W.T.; Yang, F.L.; Chiu, Y.H.; Peng, T.C.; Hsu, B.G.; Liao, K.W.; Lee, R.P. Effects of freshwater clam extract supplementation on time to exhaustion, muscle damage, pro/anti-inflammatory cytokines, and liver injury in rats after exhaustive exercise. Molecules 2013, 18, 3825–3838. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.C.; Subeq, Y.M.; Lee, C.J.; Lee, C.C.; Tsai, C.J.; Chang, F.M.; Lee, R.P. Freshwater clam extract ameliorates acute liver injury induced by hemorrhage in rats. Am. J. Chin. Med. 2008, 36, 1121–1133. [Google Scholar] [CrossRef]
- Soliman, A.M. Extract of Coelatura aegyptiaca, a freshwater clam, ameliorates hepatic oxidative stress induced by monosodium glutamate in rats. African. J. Pharmacy. Pharmacol. 2011, 5, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.P.; Subeq, Y.M.; Lee, C.J.; Hsu, B.G.; Peng, T.C. Freshwater clam extract decreased hemorrhagic shock-induced liver injury by attenuating TNF-alpha production. Biol. Res. Nurs. 2012, 14, 286–293. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Doherty, G.H. Nitric oxide in neurodegeneration: Potential benefits of non-steroidal anti-inflammatories. Neurosci. Bull. 2011, 27, 366–382. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.L.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Zuo, L.; Motherwell, M.S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 2013, 532, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Schmidt, W.J. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav. Brain Res. 2002, 136, 317–324. [Google Scholar]
- Bjorklund, A.; Kirik, D.; Rosenblad, C.; Georgievska, B.; Lundberg, C.; Mandel, R.J. Towards a neuroprotective gene therapy for Parkinson’s disease: Use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 2000, 886, 82–98. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Kordower, J.H. In vivo gene delivery of glial cell line--derived neurotrophic factor for Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S120–S132. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Rosenblad, C.; Bjorklund, A.; Mandel, R.J. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: Intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J. Neurosci. 2000, 20, 4686–4700. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef]
- Altar, C.A.; Boylan, C.B.; Jackson, C.; Hershenson, S.; Miller, J.; Wiegand, S.J.; Lindsay, R.M.; Hyman, C. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11347–11351. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.L.; Lewis, M.H.; Muzyczka, N.; Meyer, E.M. Prevention of 6-hydroxydopamine-induced rotational behavior by BDNF somatic gene transfer. Brain Res. 1999, 847, 314–320. [Google Scholar] [CrossRef]
- Gash, D.M.; Zhang, Z.; Cass, W.A.; Ovadia, A.; Simmerman, L.; Martin, D.; Russell, D.; Collins, F.; Hoffer, B.J.; Gerhardt, G.A. Morphological and functional effects of intranigrally administered GDNF in normal rhesus monkeys. J. Comp. Neurol. 1995, 363, 345–358. [Google Scholar] [CrossRef]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef]
- Eslamboli, A.; Georgievska, B.; Ridley, R.M.; Baker, H.F.; Muzyczka, N.; Burger, C.; Mandel, R.J.; Annett, L.; Kirik, D. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson’s disease. J. Neurosci. 2005, 25, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, Y.; Lin, Q.; Collier, T.J.; Frim, D.M.; Breakefield, X.O.; Bohn, M.C. Astrocytes retrovirally transduced with BDNF elicit behavioral improvement in a rat model of Parkinson’s disease. Brain Res. 1995, 691, 25–36. [Google Scholar] [CrossRef]
- Levivier, M.; Przedborski, S.; Bencsics, C.; Kang, U.J. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1995, 15, 7810–7820. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.T.; Lee, J.Y.; Lee, J.; Kim, H.; Yoon, K.S.; Choe, W.; Kang, I. Oleic acid reduces lipopolysaccharide-induced expression of iNOS and COX-2 in BV2 murine microglial cells: Possible involvement of reactive oxygen species, p38 MAPK, and IKK/NF-kappaB signaling pathways. Neurosci. Lett. 2009, 464, 93–97. [Google Scholar] [CrossRef]
- Guo, X.; Li, H.; Xu, H.; Halim, V.; Zhang, W.; Wang, H.; Ong, K.T.; Woo, S.L.; Walzem, R.L.; Mashek, D.G.; et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS ONE 2012, 7, e39286. [Google Scholar] [CrossRef]
- Patel, N.K.; Khan, M.S.; Bhutani, K.K. Investigations on Leucas cephalotes (Roth.) Spreng. for inhibition of LPS-induced pro-inflammatory mediators in murine macrophages and in rat model. EXCLI. J 2015, 14, 508–516. [Google Scholar]
- Montserrat-de la, P.S.; Fernandez-Arche, Á.; Ángel-Martin, M.; Garcia-Gimenez, M.D. The sterols isolated from Evening Primrose oil modulate the release of proinflammatory mediators. Phytomedicine 2012, 19, 1072–1076. [Google Scholar] [CrossRef]
- Kung, W.M.; Lin, M.S. The NFkB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 934. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Target | Sequence (5’ -> 3’) | Product Size (bp) | Sequence Reference | |
|---|---|---|---|---|
| Actin | forward | GCTACAGCTTCACCACCACA | 123 | NM_007393.5 |
| reverse | TCTCCAGGGAGGAAGAGGAT | |||
| BDNF | forward | TGGCTGACACTTTTGAGCAC | 131 | NM_001316310.1 |
| reverse | CAAAGGCACTTGACTGCTGA | |||
| GDNF | forward | TGGGCTATGAAACCAAGGAG | 142 | NM_001301357.1 |
| reverse | CAACATGCCTGGCCTACTTT | |||
| TNF-α | forward | CAGGGGCCACCACGCTCTTC | 371 | NM_001278601.1 |
| reverse | CTTGGGGCAGGGGCTCTTGAC | |||
| IL-1β | forward | CAGGCTCCGAGATGAACAACAAAA | 332 | NM_008361.4 |
| reverse | TGGGGAACTCTGCAGACTCAAACT | |||
| iNOS | forward | TCACTGGGACAGCACAGAAT | 510 | NM_001313922.1 |
| reverse | TGTGTCTGCAGATGTGCTGA | |||
| GPx | forward | CCTCAAGTACGTCCGGCCTG | 197 | NM_008160.6 |
| reverse | CAACATCGTTGCGACACACC | |||
| SOD | forward | TGGGTTCCACGTCCATCAGTA | 151 | NM_011434.1 |
| reverse | ACCGTCCTTTCCAGCAGTCA | |||
| Catalase | forward | TTCAGAAGAAAGCGGTCAAGAAT | 59 | NM_009804.2 |
| reverse | GATGCGGGCCCCATAGTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.-S.; Chen, S.-M.; Hua, K.-F.; Chen, W.-J.; Hsieh, C.-C.; Lin, C.-C. Freshwater Clam Extract Mitigates Neuroinflammation and Amplifies Neurotrophic Activity of Glia: Insights from In Vitro Model of Neurodegenerative Pathomechanism. J. Clin. Med. 2022, 11, 553. https://doi.org/10.3390/jcm11030553
Lin M-S, Chen S-M, Hua K-F, Chen W-J, Hsieh C-C, Lin C-C. Freshwater Clam Extract Mitigates Neuroinflammation and Amplifies Neurotrophic Activity of Glia: Insights from In Vitro Model of Neurodegenerative Pathomechanism. Journal of Clinical Medicine. 2022; 11(3):553. https://doi.org/10.3390/jcm11030553
Chicago/Turabian StyleLin, Muh-Shi, Shu-Mei Chen, Kuo-Feng Hua, Wei-Jung Chen, Cho-Chen Hsieh, and Chai-Ching Lin. 2022. "Freshwater Clam Extract Mitigates Neuroinflammation and Amplifies Neurotrophic Activity of Glia: Insights from In Vitro Model of Neurodegenerative Pathomechanism" Journal of Clinical Medicine 11, no. 3: 553. https://doi.org/10.3390/jcm11030553