
Endothelial Dysfunction after Hematopoietic Stem Cell Transplantation: A Review Based on Physiopathology

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
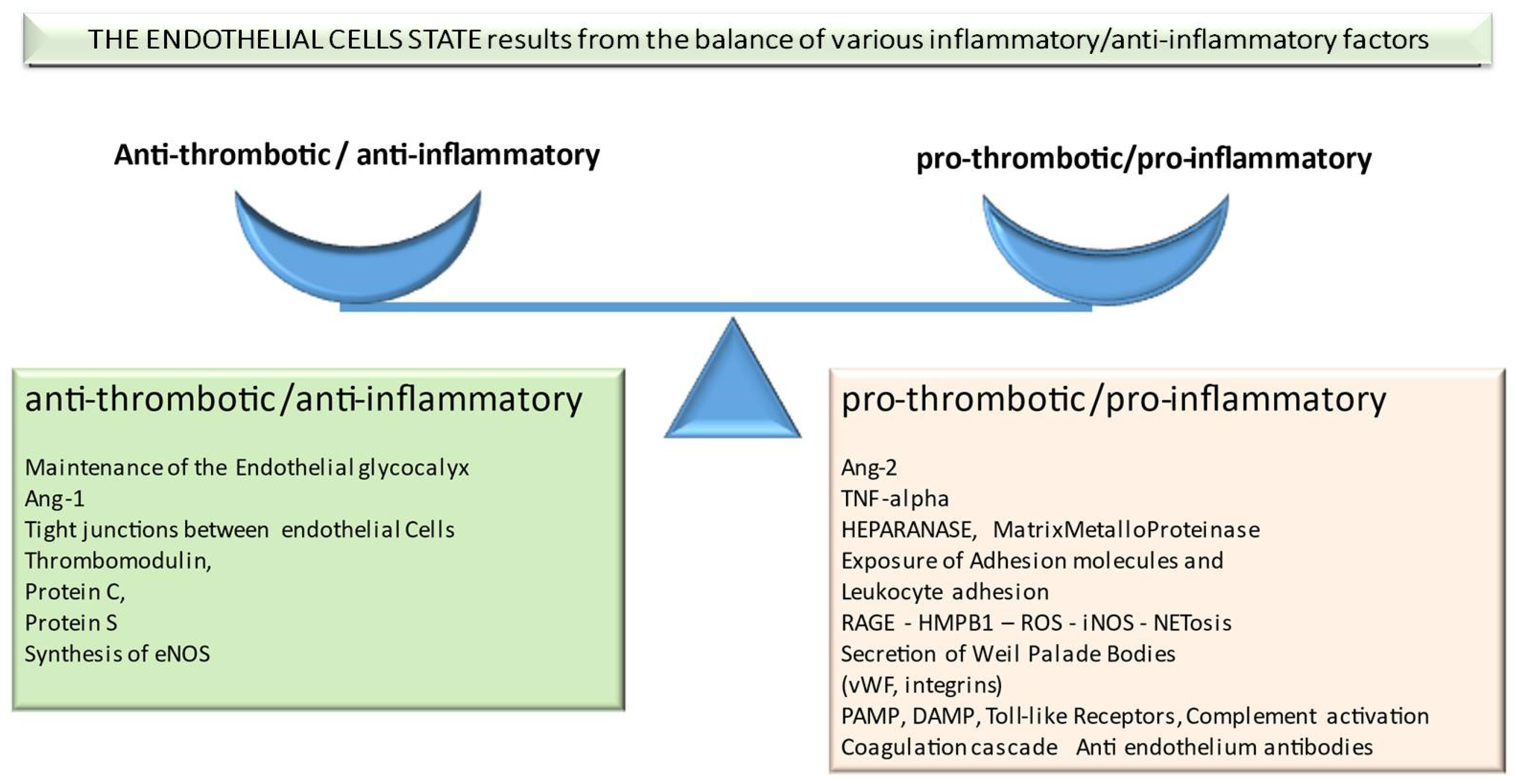
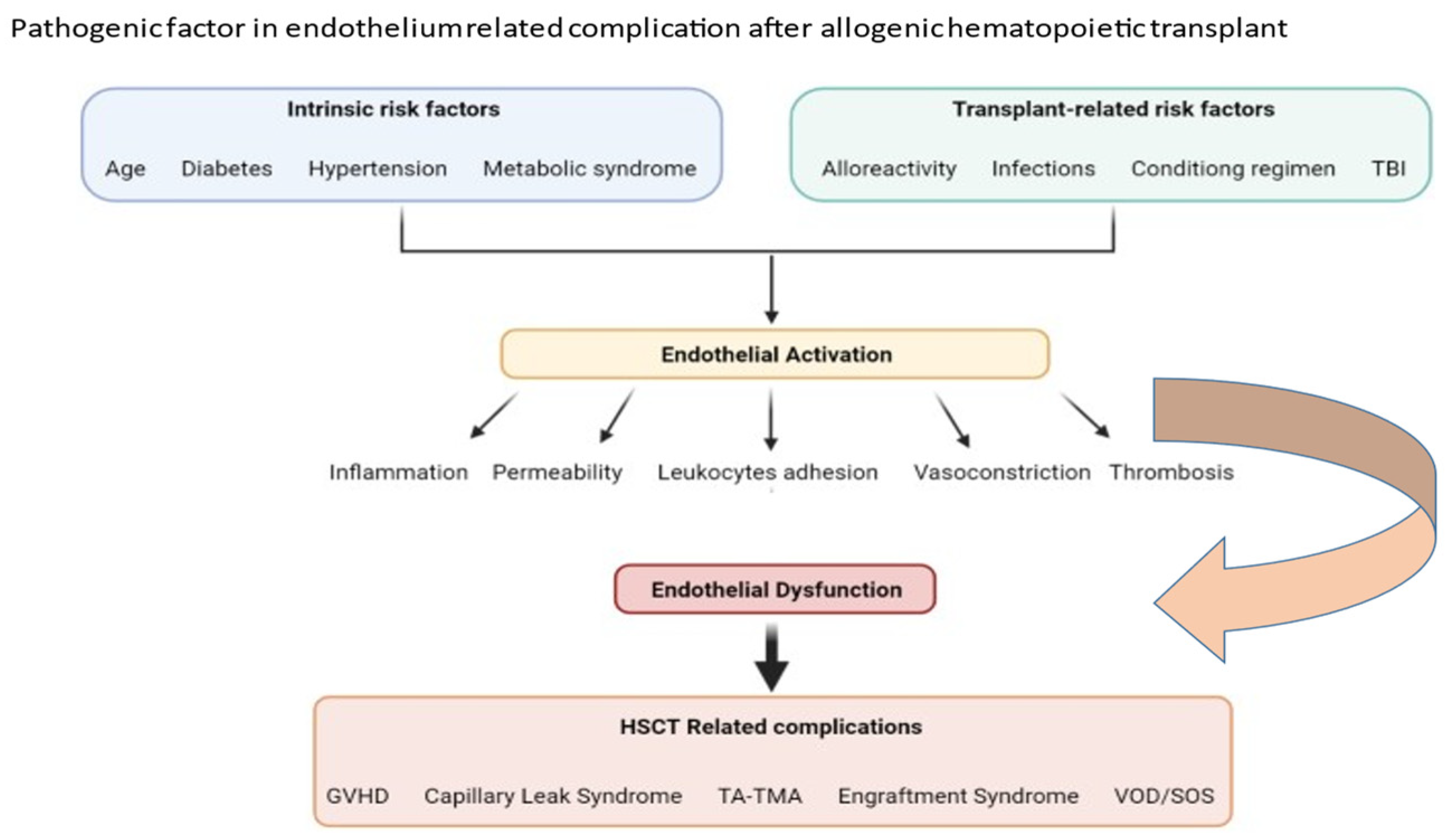
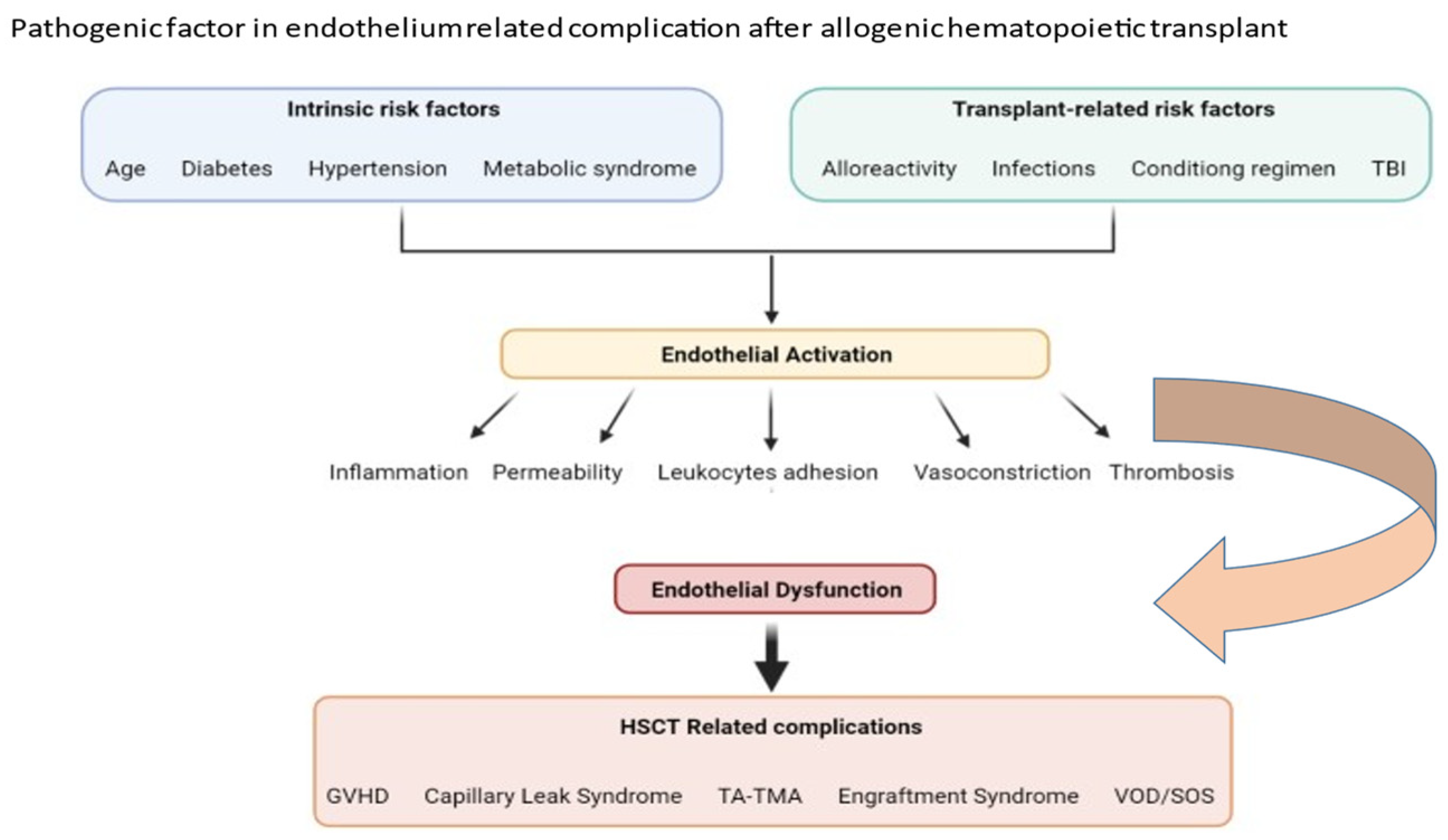
2. Factors Important in Pathophysiology of Transplant-Associated Endothelium Dysfunction
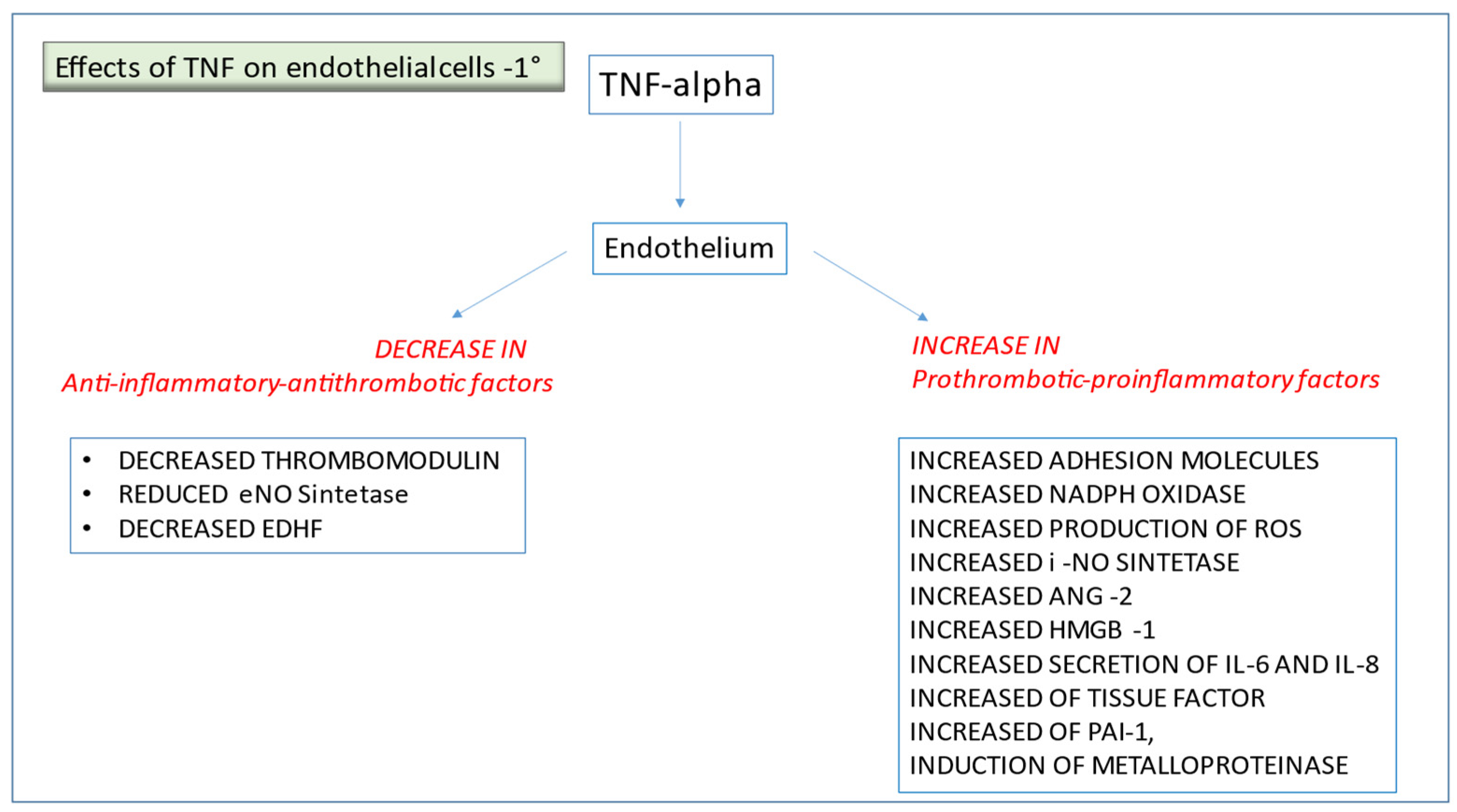
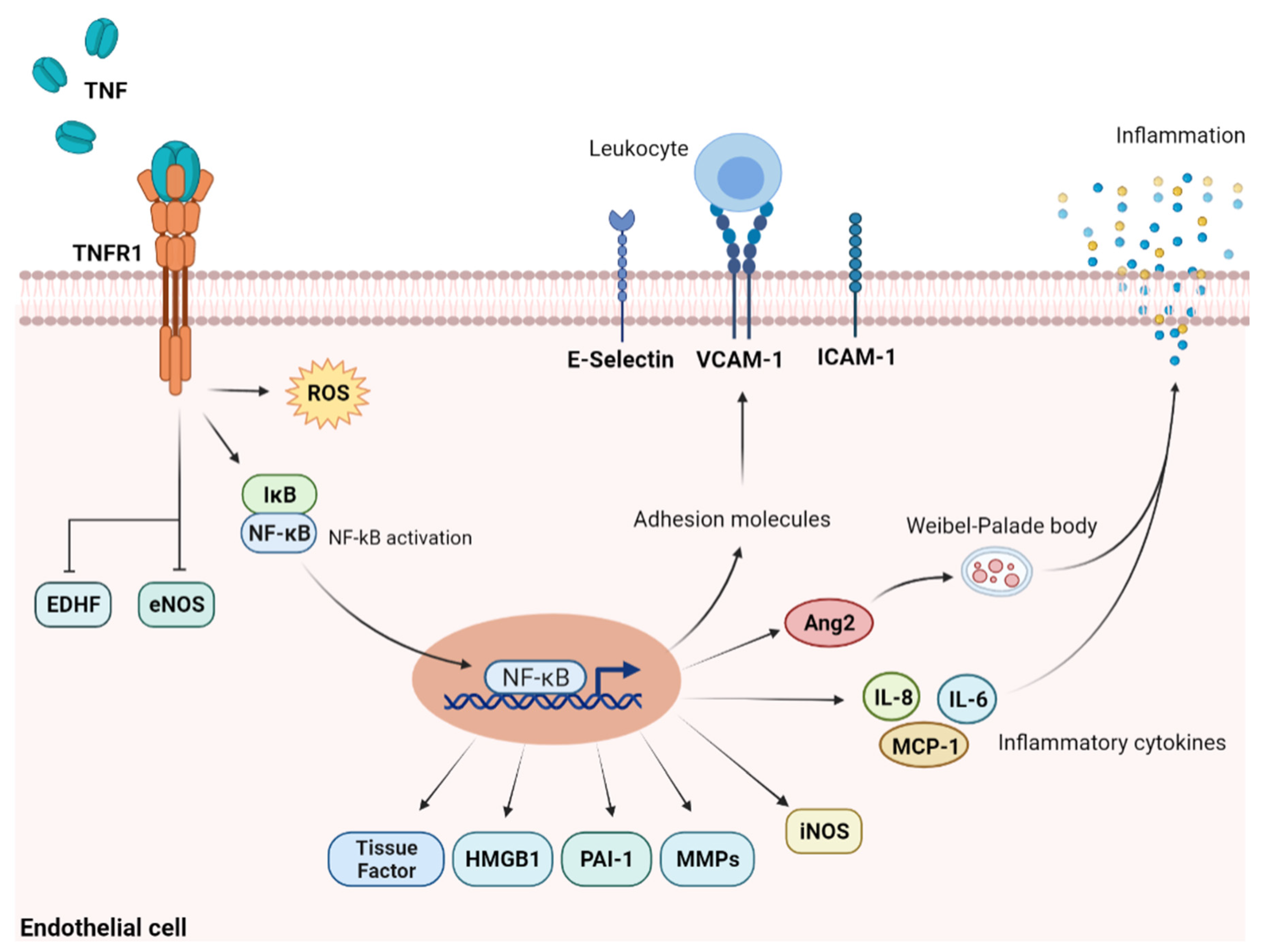
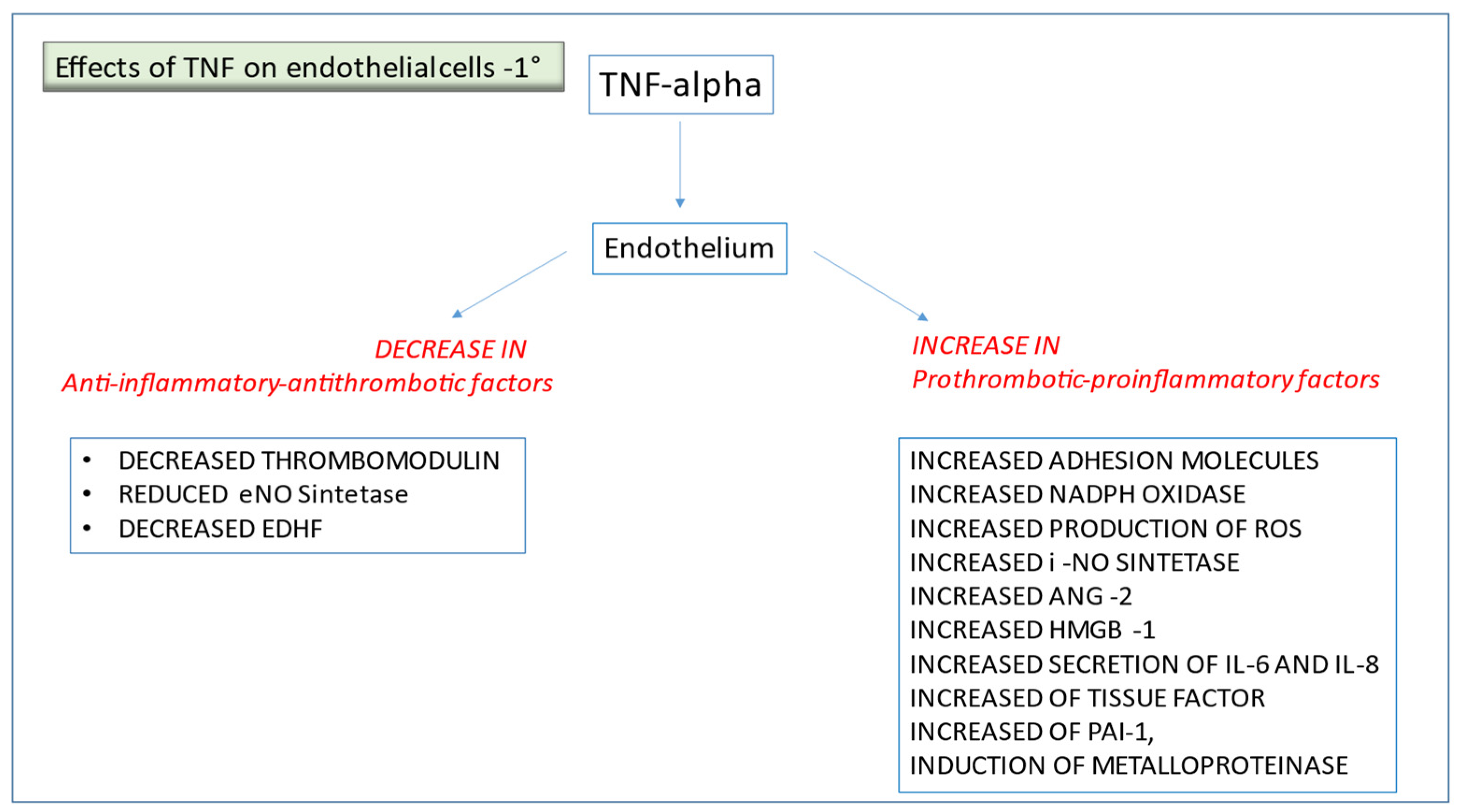
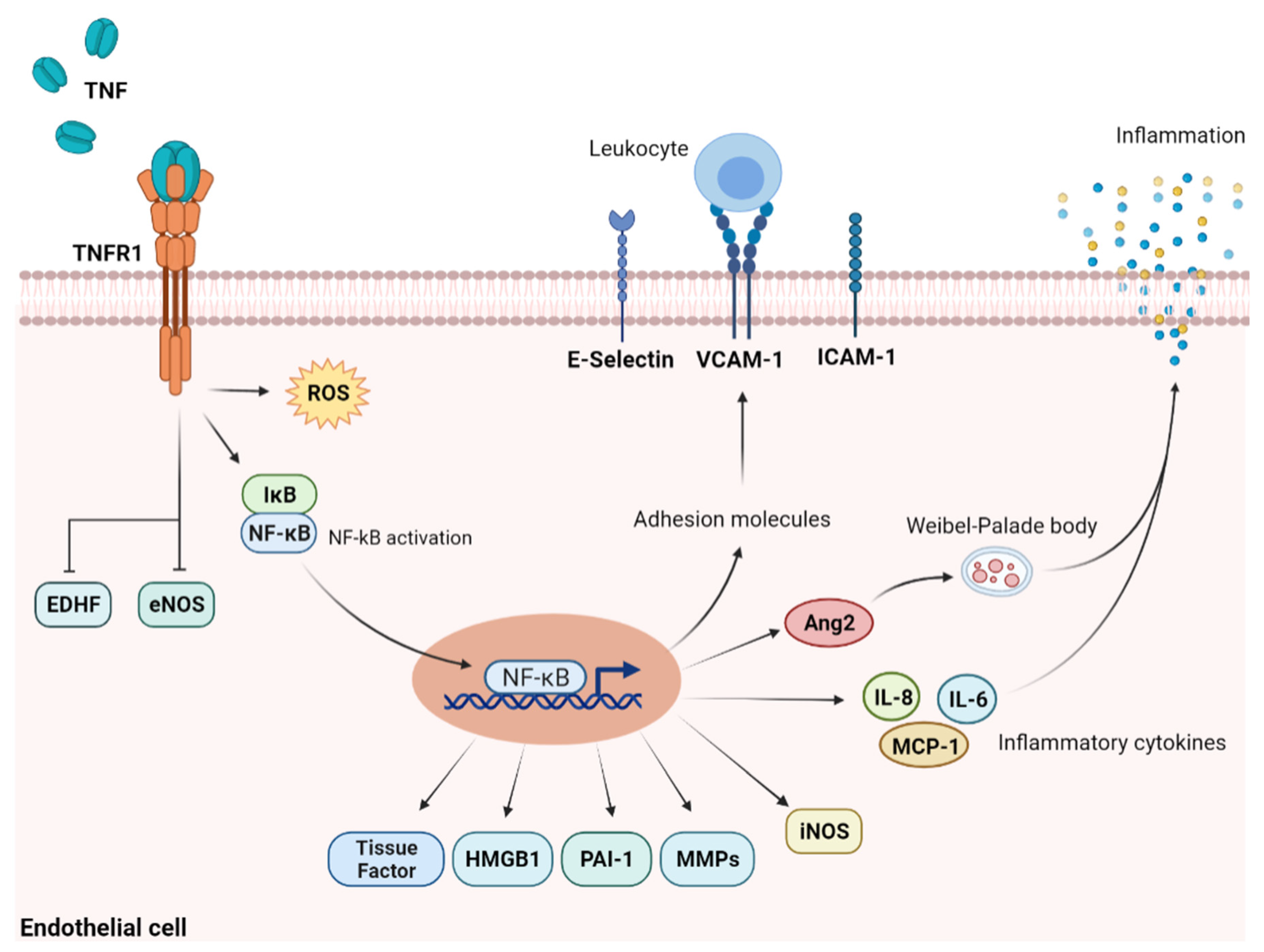
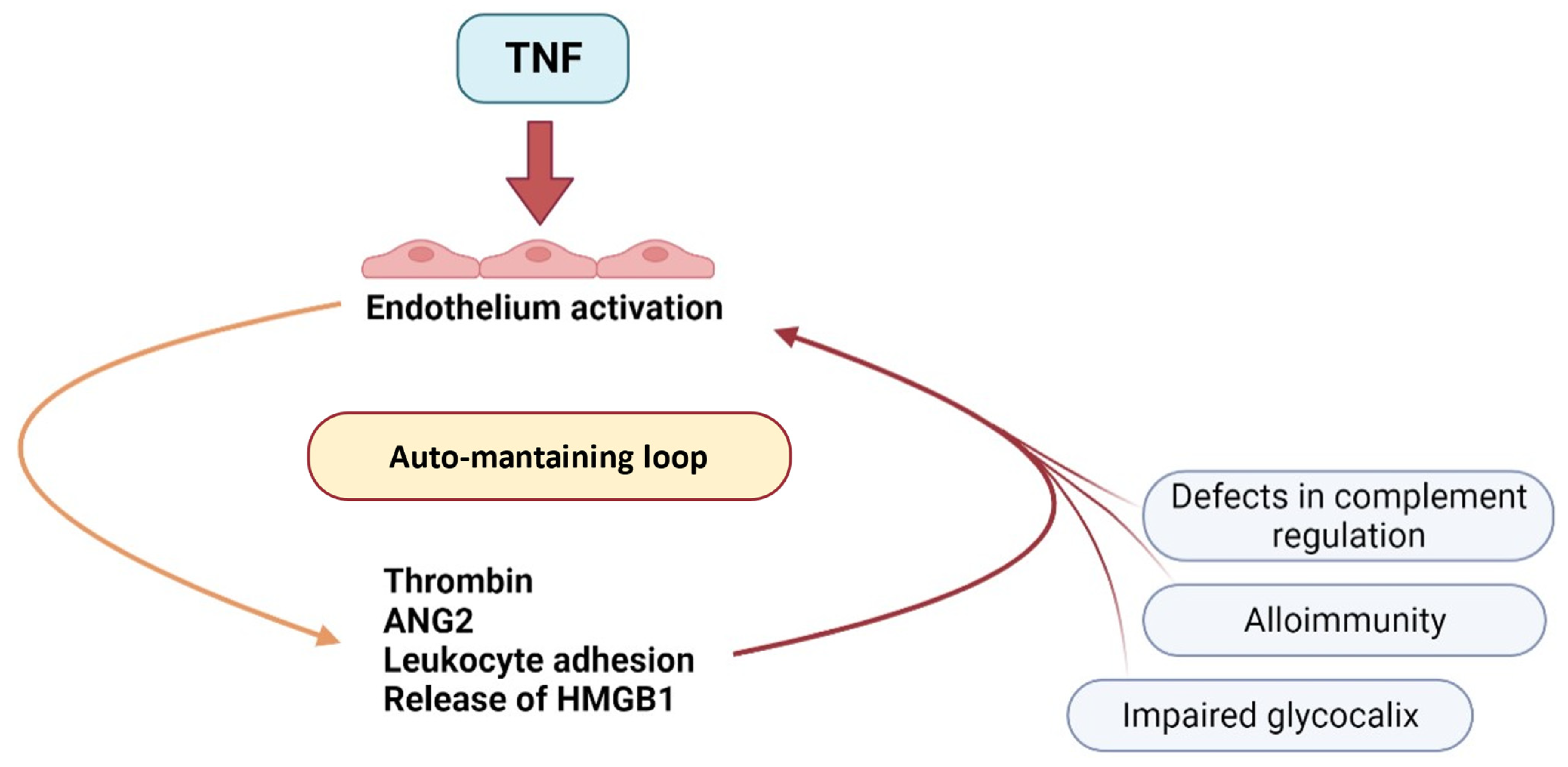
2.1. TNF-α and Other Pro-Inflammatory Cytokines
2.2. Engagement of Adhesion Molecules and Toll-like Receptors
2.3. Endothelium Activation by Antibodies
2.4. Cadherin
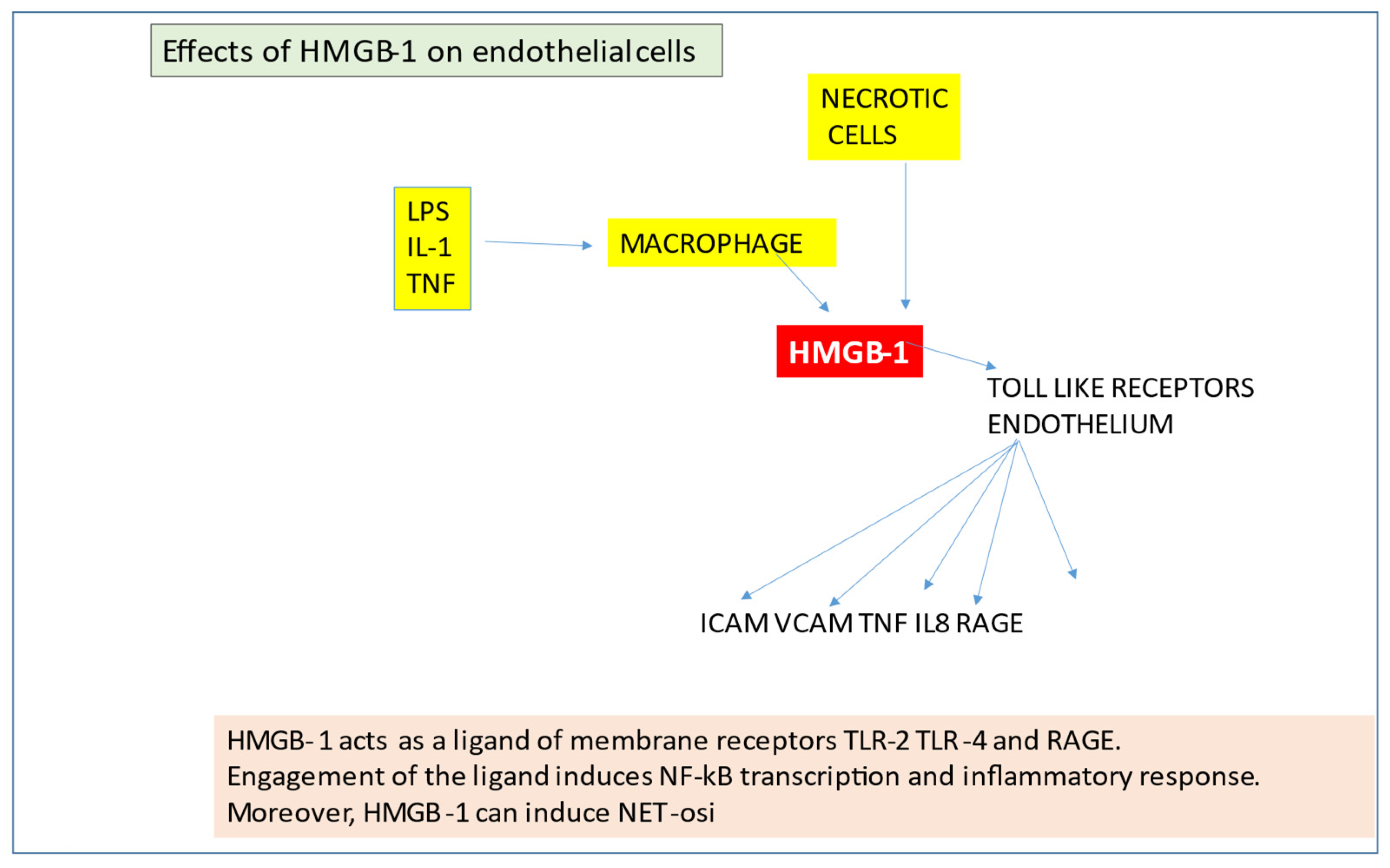
2.5. High-Mobility Group B-1
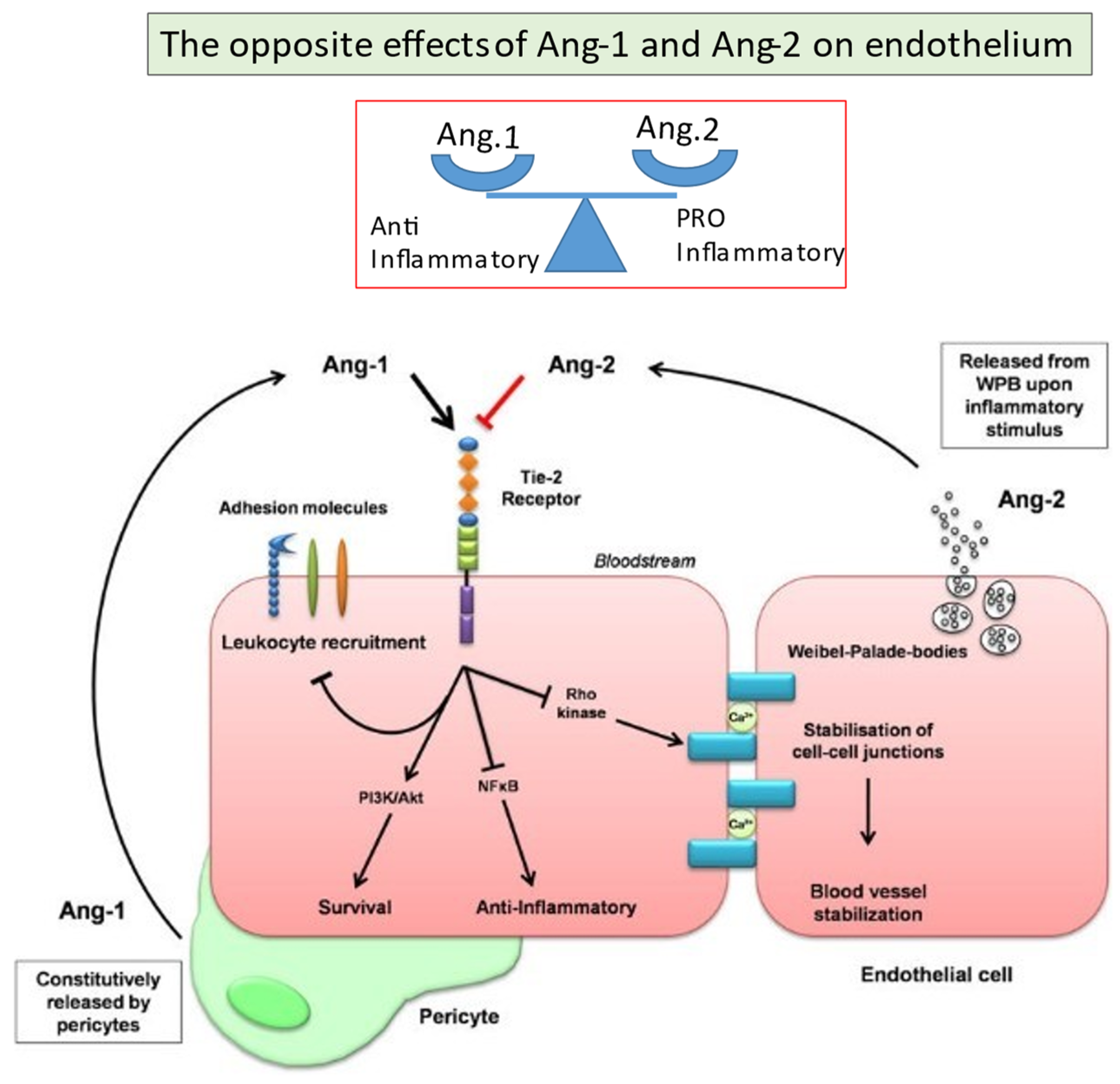
2.6. Angiopoietins
2.7. Coagulation Cascade
2.8. Nitric Oxide
2.9. Glycocalyx
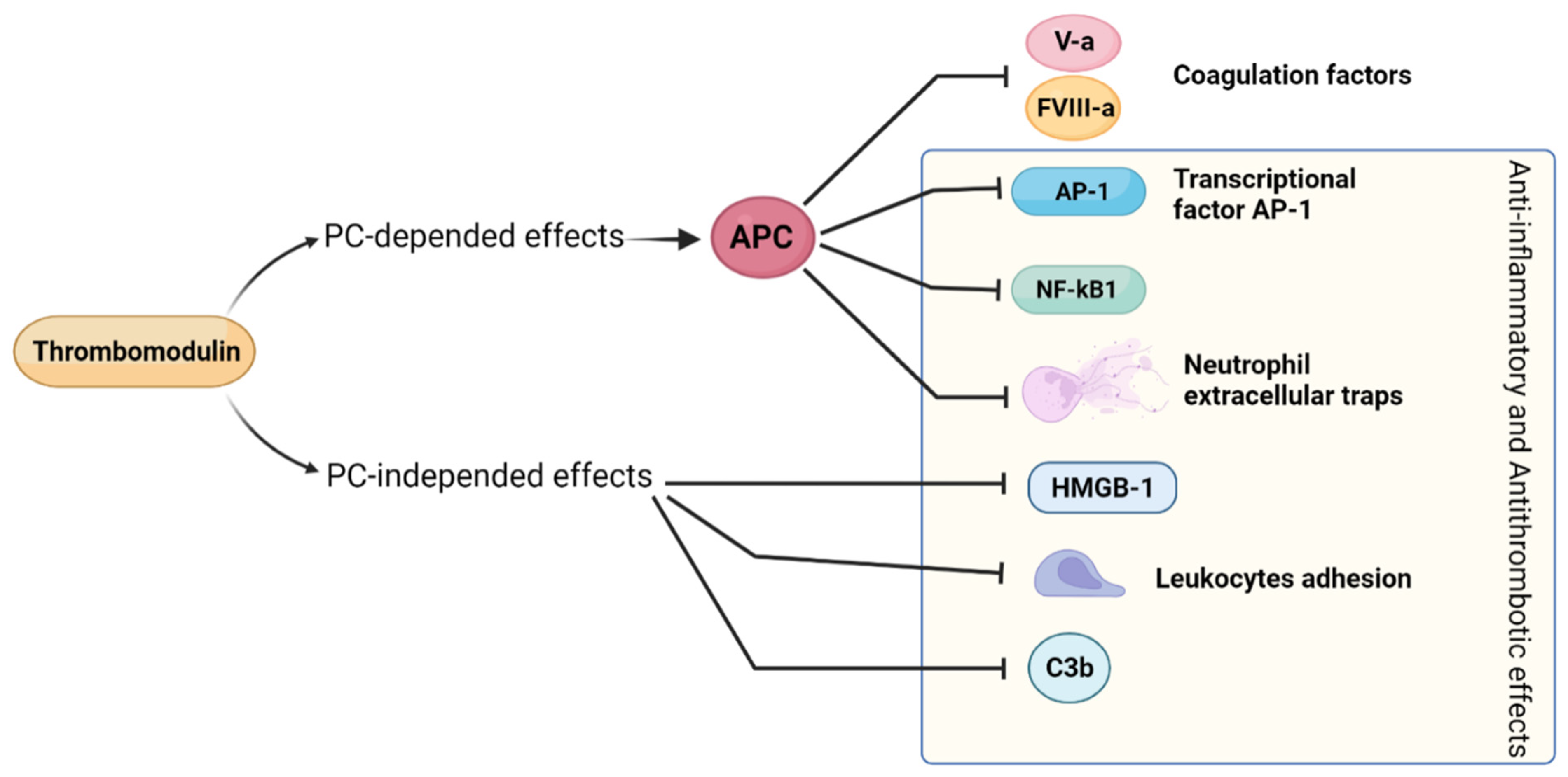
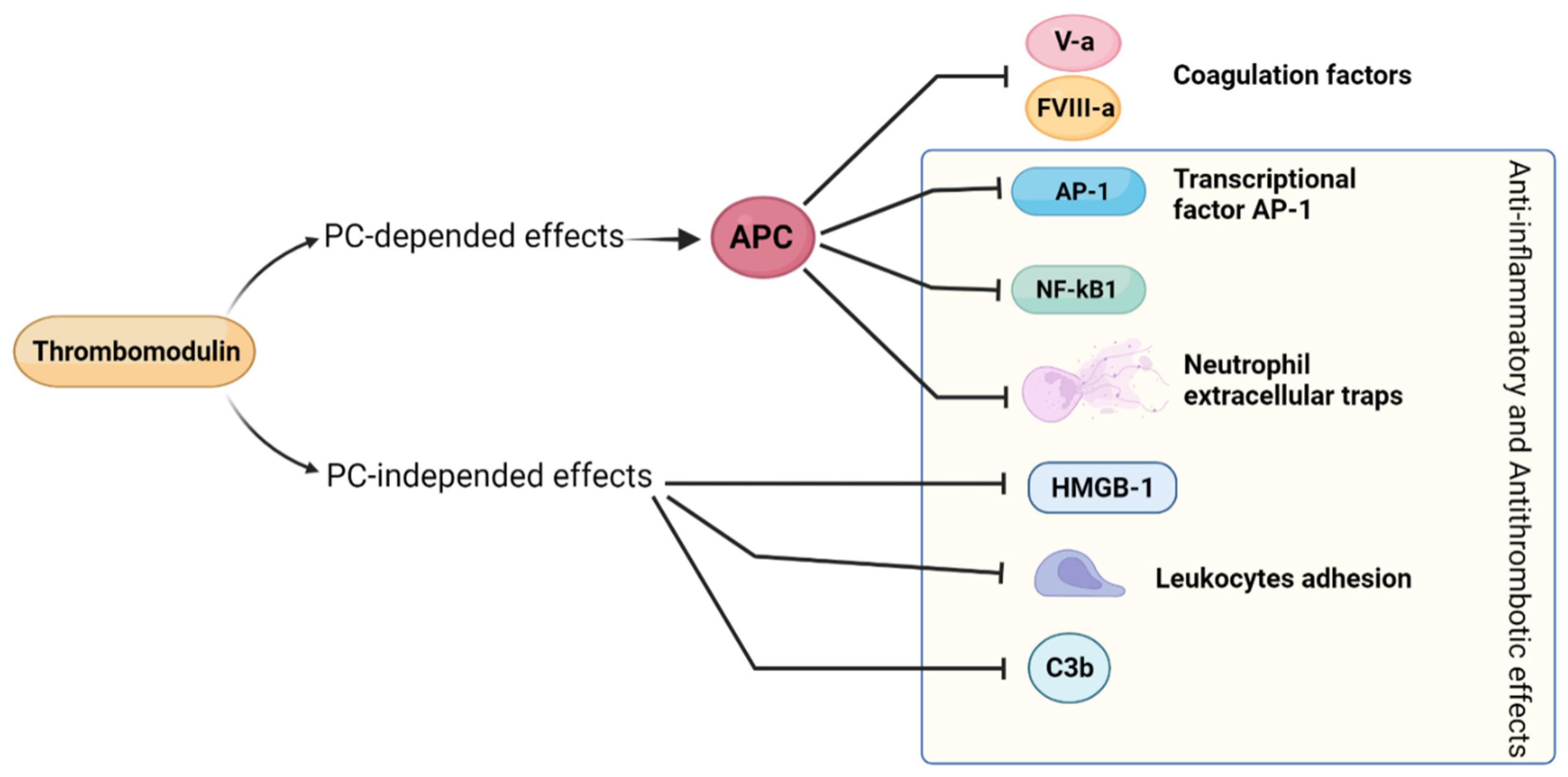
2.10. Thrombomodulin
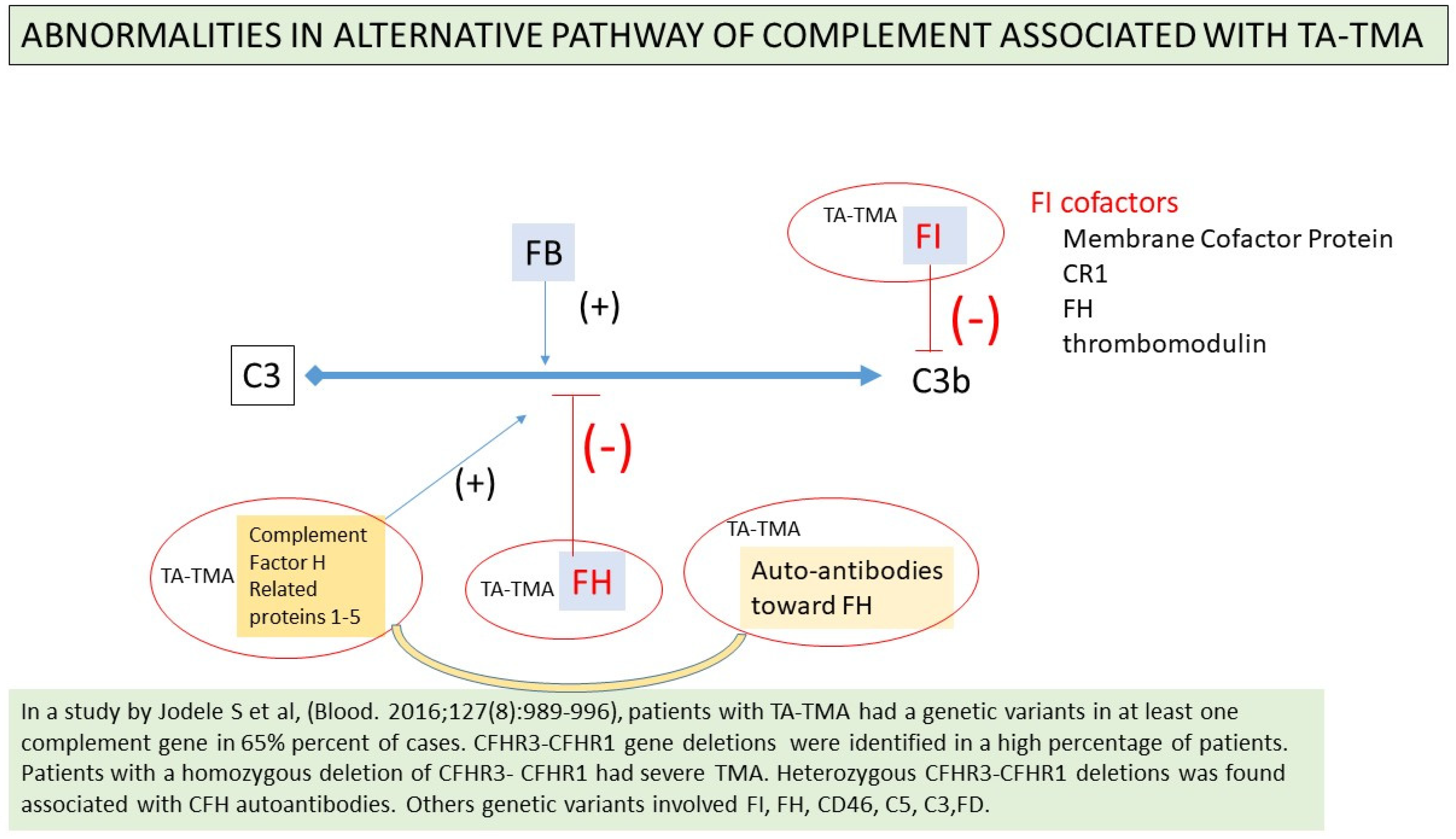
2.11. Complement Activation
2.12. Neutrophil Extracellular Trap (NET)
2.13. Endothelin-1
2.14. Microvesicles
3. Clinical Pictures of Endothelial Dysfunction after Allogeneic HSCT
3.1. SOS/VOD
3.2. Capillary Leak Syndrome (CLS)
3.3. Idiopathic Pneumonia Syndrome (IPS)
3.4. Engraftment Syndrome (ES)
3.5. Transplant-Associated Thrombotic Microangiopathy (TA-TMA)
3.6. Endothelial Dysfunction and GVHD
4. Therapeutic Interventions for EC Dysfunction
4.1. Defibrotide
4.2. Anti-complement Agents
4.3. Anti-CD20 (Rituximab)
4.4. Withdrawal of Calcineurin Inhibitors
4.5. Therapeutic Plasma Exchange
4.6. Thrombomodulin
4.7. Statins
4.8. Angiopoietin1 Mimetics
4.9. Alpha-1Anti-Trypsin (A1AT)
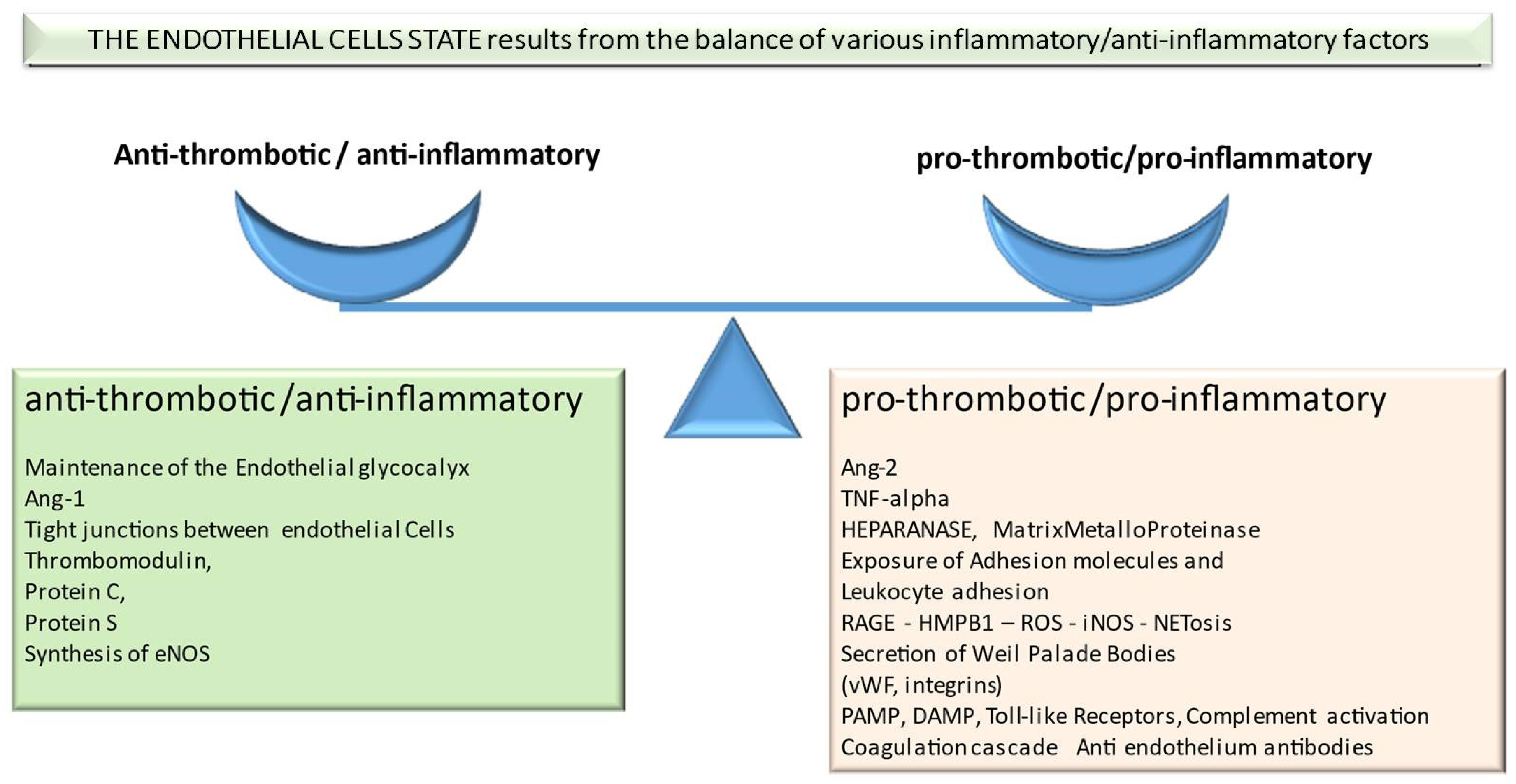
5. Conclusions and Working Hypothesis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- De Backer, D.; Orbegozo Cortes, D.; Donadello, K.; Vincent, J.-L. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence 2014, 5, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [Green Version]
- Penack, O.; Luft, T. Editorial: Endothelial Dysfunction During Inflammation and Alloimmunity. Front. Immunol. 2018, 9, 2886. [Google Scholar] [CrossRef]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens. Res. 2012, 35, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; De Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Lia, G.; Giaccone, L.; Leone, S.; Bruno, B. Biomarkers for Early Complications of Endothelial Origin After Allogeneic Hematopoietic Stem Cell Transplantation: Do They Have a Potential Clinical Role? Front. Immunol. 2021, 12, 1869. [Google Scholar] [CrossRef]
- Carreras, E.; Diaz-Ricart, M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant. 2011, 46, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Suitters, A.; Rose, M.; Higgins, A.; Yacoub, M.H. MHC antigen expression in sequential biopsies from cardiac transplant patients--correlation with rejection. Clin. Exp. Immunol. 1987, 69, 575–583. [Google Scholar]
- Maenaka, A.; Kenta, I.; Ota, A.; Miwa, Y.; Ohashi, W.; Horimi, K.; Matsuoka, Y.; Ohnishi, M.; Uchida, K.; Kobayashi, T. Interferon-γ-induced HLA Class II expression on endothelial cells is decreased by inhibition of mTOR and HMG-CoA reductase. FEBS Open Bio 2020, 10, 927–936. [Google Scholar] [CrossRef] [Green Version]
- González-Molina, M.; Ruiz-Esteban, P.; Caballero, A.; Burgos, D.; Cabello, M.; Leon, M.; Fuentes, L.; Hernandez, D. Immune response and histology of humoral rejection in kidney transplantation. Nefrologia 2016, 36, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Loupy, A.; Haas, M.; Solez, K.; Racusen, L.; Glotz, D.; Seron, D.; Nankivell, B.J.; Colvin, R.B.; Afrouzian, M.; Akalin, E.; et al. The Banff 2015 Kidney Meeting Report: Current Challenges in Rejection Classification and Prospects for Adopting Molecular Pathology. Am. J. Transplant. 2017, 17, 28–41. [Google Scholar] [CrossRef]
- Hammond, M.E.H.; Revelo, M.P.; Miller, D.V.; Snow, G.L.; Budge, D.; Stehlik, J.; Molina, K.M.; Selzman, C.H.; Drakos, S.G.; Rami, A.A.; et al. ISHLT pathology antibody mediated rejection score correlates with increased risk of cardiovascular mortality: A retrospective validation analysis. J. Heart Lung Transplant. 2016, 35, 320–325. [Google Scholar] [CrossRef]
- Zhang, X.; Reed, E.F. Effect of Antibodies on Endothelium. Am. J. Transplant. 2009, 9, 2459–2465. [Google Scholar] [CrossRef]
- Biedermann, B.C.; Sahner, S.; Gregor, M.; Tsakiris, D.A.; Jeanneret, C.; Pober, J.S.; Gratwohl, A. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet 2002, 359, 2078–2083. [Google Scholar] [CrossRef]
- Biedermann, B.C. Vascular endothelium and graft-versus-host disease. Best Pract. Res. Clin. Haematol. 2008, 21, 129–138. [Google Scholar] [CrossRef]
- Maeda, Y. Pathogenesis of graft-versus-host disease: Innate immunity amplifying acute alloimmune responses. Int. J. Hematol. 2013, 98, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Luft, T.; Dreger, P.; Radujkovic, A. Endothelial cell dysfunction: A key determinant for the outcome of allogeneic stem cell transplantation. Bone Marrow Transplant. 2021, 56, 2326–2335. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Vassalli, P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992, 10, 411–452. [Google Scholar] [CrossRef]
- Swiatkowska, M.; Szemraj, J.; Cierniewski, C.S. Induction of PAI-1 expression by tumor necrosis factor α in endothelial cells is mediated by its responsive element located in the 4G/5G site. FEBS J. 2005, 272, 5821–5831. [Google Scholar] [CrossRef]
- Zhou, L.; Yan, C.; Gieling, R.G.; Kida, Y.; Garner, W.; Li, W.; Han, Y.-P. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated Kinase-1. BMC Immunol. 2009, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Han, N.J.; Kim, J.J.; Lee, M.J.; Park, S.-K. TNF-α Activates High-Mobility Group Box 1-Toll-Like Receptor 4 Signaling Pathway in Human Aortic Endothelial Cells. Cell. Physiol. Biochem. 2016, 38, 2139–2151. [Google Scholar] [CrossRef]
- Chen, X.; Andresen1, B.T.; Hill, M.; Zhang, J.; Booth, F.; Zhang, C. Role of Reactive Oxygen Species in Tumor Necrosis Factor-alpha Induced Endothelial Dysfunction. Curr. Hypertens. Rev. 2008, 4, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-M.; Fan, L.M.; Christie, M.R.; Shah, A.M. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 2005, 25, 2320–2330. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pelekanakis, K.; Woolkalis, M.J. Thrombin and tumor necrosis factor alpha synergistically stimulate tissue factor expression in human endothelial cells: Regulation through c-Fos and c-Jun. J. Biol. Chem. 2004, 279, 36142–36147. [Google Scholar] [CrossRef] [Green Version]
- Holler, E.; Kolb, H.J.; Möller, A.; Kempeni, J.; Liesenfeld, S.; Pechumer, H.; Lehmacher, W.; Ruckdeschel, G.; Gleixner, B.; Riedner, C. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood 1990, 75, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Symington, F.W.; Pepe, M.S.; Chen, A.B.; Deliganis, A. Serum tumor necrosis factor alpha associated with acute graft-versus-host disease in humans. Transplantation 1990, 50, 518–521. [Google Scholar] [CrossRef]
- Muller, W.A.; Weigl, S.A.; Deng, X.; Phillips, D.M. PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 1993, 178, 449–460. [Google Scholar] [CrossRef]
- Kanters, E.; van Rijssel, J.; Hensbergen, P.J.; Hondius, D.; Mul, F.P.J.; Deelder, A.M.; Sonnenberg, A.; van Buul, J.D.; Hordijk, P.L. Filamin B Mediates ICAM-1-driven Leukocyte Transendothelial Migration. J. Biol. Chem. 2008, 283, 31830–31839. [Google Scholar] [CrossRef] [Green Version]
- Gloude, N.J.; Khandelwal, P.; Luebbering, N.; Lounder, D.T.; Jodele, S.; Alder, M.N.; Lane, A.; Wilkey, A.; Lake, K.E.; Litts, B.; et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood 2017, 130, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Cary, L.A.; Guan, J.L. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 1999, 4, D102-13. [Google Scholar] [CrossRef] [Green Version]
- Kornblit, B.; Müller, K. Sensing danger: Toll-like receptors and outcome in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Bhagwani, A.; Thompson, A.A.R.; Farkas, L. When Innate Immunity Meets Angiogenesis—The Role of Toll-Like Receptors in Endothelial Cells and Pulmonary Hypertension. Front. Med. 2020, 7, 352. [Google Scholar] [CrossRef]
- Wehner, J.; Morrell, C.N.; Reynolds, T.; Rodriguez, E.R.; Baldwin, W.M. Antibody and complement in transplant vasculopathy. Circ. Res. 2007, 100, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.-P.; Korin, Y.; Zhang, X.; Jindra, P.T.; Rozengurt, E.; Reed, E.F. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J. Immunol. 2007, 178, 7911–7922. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.H.; Platt, J.L. Accommodation of grafts: Implications for health and disease. Hum. Immunol. 2007, 68, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Belin, C.; Devic, P.; Ayrignac, X.; Dos Santos, A.; Paix, A.; Sirven-Villaros, L.; Simard, C.; Lamure, S.; Gastinne, T.; Ursu, R.; et al. Description of neurotoxicity in a series of patients treated with CAR T-cell therapy. Sci. Rep. 2020, 10, 18997. [Google Scholar] [CrossRef]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell-Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.-T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Ghosh, C.C.; Patel, R.; Iwaki, S.; Gaskins, D.; Nelson, C.; Jones, N.; Greipp, P.R.; Parikh, S.M.; Druey, K.M. Vascular endothelial hyperpermeability induces the clinical symptoms of Clarkson disease (the systemic capillary leak syndrome). Blood 2012, 119, 4321–4332. [Google Scholar] [CrossRef]
- Duan, L.; Wang, C.-Y.; Chen, J.; Gong, Q.; Zhu, P.; Zheng, F.; Tan, Z.; Gong, F.; Fang, M. High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab. Investig. 2011, 91, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.N.; Tozawa, Y.; Deutsch, U.; Wolburg-Buchholz, K.; Fujiwara, Y.; Gendron-Maguire, M.; Gridley, T.; Wolburg, H.; Risau, W.; Qin, Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376, 70–74. [Google Scholar] [CrossRef]
- Jones, N.; Chen, S.H.; Sturk, C.; Master, Z.; Tran, J.; Kerbel, R.S.; Dumont, D.J. A unique autophosphorylation site on Tie2/Tek mediates Dok-R phosphotyrosine binding domain binding and function. Mol. Cell. Biol. 2003, 23, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Dumont, D.J.; Gradwohl, G.; Fong, G.H.; Puri, M.C.; Gertsenstein, M.; Auerbach, A.; Breitman, M.L. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994, 8, 1897–1909. [Google Scholar] [CrossRef] [Green Version]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Pizurki, L.; Zhou, Z.; Glynos, K.; Roussos, C.; Papapetropoulos, A. Angiopoietin-1 inhibits endothelial permeability, neutrophil adherence and IL-8 production. Br. J. Pharmacol. 2003, 139, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 2000, 6, 460–463. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Gamble, J.R.; Drew, J.; Trezise, L.; Underwood, A.; Parsons, M.; Kasminkas, L.; Rudge, J.; Yancopoulos, G.; Vadas, M.A. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ. Res. 2000, 87, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Moon, S.O.; Park, S.K.; Chae, S.W.; Koh, G.Y. Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ. Res. 2001, 89, 477–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Fulton, D.; Mahboubi, K.; Kalb, R.G.; O’Connor, D.S.; Li, F.; Altieri, D.C.; Sessa, W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000, 275, 9102–9105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Kim, H.G.; So, J.N.; Kim, J.H.; Kwak, H.J.; Koh, G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-Kinase/Akt signal transduction pathway. Circ. Res. 2000, 86, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBusk, L.M.; Hallahan, D.E.; Lin, P.C. Akt is a major angiogenic mediator downstream of the Ang1/Tie2 signaling pathway. Exp. Cell Res. 2004, 298, 167–177. [Google Scholar] [CrossRef]
- Ziegler, T.; Horstkotte, J.; Schwab, C.; Pfetsch, V.; Weinmann, K.; Dietzel, S.; Rohwedder, I.; Hinkel, R.; Gross, L.; Lee, S.; et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J. Clin. Investig. 2013, 123, 3436–3445. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, M.; van Nieuw Amerongen, G.P.; van Bezu, J.; Paul, M.A.; Groeneveld, A.B.J.; van Hinsbergh, V.W.M. Opposing effects of the angiopoietins on the thrombin-induced permeability of human pulmonary microvascular endothelial cells. PLoS ONE 2011, 6, e23448. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef]
- Rathnakumar, K.; Savant, S.; Giri, H.; Ghosh, A.; Fisslthaler, B.; Fleming, I.; Ram, U.; Bera, A.K.; Augustin, H.G.; Dixit, M. Angiopoietin-2 mediates thrombin-induced monocyte adhesion and endothelial permeability. J. Thromb. Haemost. 2016, 14, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Sauthoff, H.; Herscovici, P.; Pipiya, T.; Cheng, J.; Heitner, S.; Szentirmai, O.; Carter, B.; Hay, J.G. Angiopoietin-1 increases survival and reduces the development of lung edema induced by endotoxin administration in a murine model of acute lung injury. Crit. Care Med. 2008, 36, 262–267. [Google Scholar] [CrossRef]
- Cai, J.; Kehoe, O.; Smith, G.M.; Hykin, P.; Boulton, M.E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.L.; Jarvis, J.; Jennings, I.; Luddington, R.; Marcus, R.E. Changes in the natural anticoagulants following bone marrow transplantation. Bone Marrow Transplant. 1990, 5, 39–42. [Google Scholar] [PubMed]
- Gordon, B.; Haire, W.; Kessinger, A.; Duggan, M.; Armitage, J. High frequency of antithrombin 3 and protein C deficiency following autologous bone marrow transplantation for lymphoma. Bone Marrow Transplant. 1991, 8, 497–502. [Google Scholar] [PubMed]
- Collins, P.W.; Gutteridge, C.N.; O’Driscoll, A.; Blair, S.; Jones, L.; Aitchison, R.; Kelsey, S.M.; Chopra, R.; Goldstone, A.; Newland, A.C. von Willebrand factor as a marker of endothelial cell activation following BMT. Bone Marrow Transplant. 1992, 10, 499–506. [Google Scholar]
- Catani, L.; Gugliotta, L.; Mattioli Belmonte, M.; Vianelli, N.; Gherlinzoni, F.; Miggiano, M.C.; Belardinelli, A.R.; Rosti, G.; Calori, E.; Bandini, G. Hypercoagulability in patients undergoing autologous or allogeneic BMT for hematological malignancies. Bone Marrow Transplant. 1993, 12, 253–259. [Google Scholar]
- Salat, C.; Holler, E.; Reinhardt, B.; Kolb, H.J.; Seeber, B.; Ledderose, G.; Mittermueller, J.; Duell, T.; Wilmanns, W.; Hiller, E. Parameters of the fibrinolytic system in patients undergoing BMT: Elevation of PAI-1 in veno-occlusive disease. Bone Marrow Transplant. 1994, 14, 747–750. [Google Scholar]
- Park, Y.D.; Yasui, M.; Yoshimoto, T.; Chayama, K.; Shimono, T.; Okamura, T.; Inoue, M.; Yumura-Yagi, K.; Kawa-Ha, K. Changes in hemostatic parameters in hepatic veno-occlusive disease following bone marrow transplantation. Bone Marrow Transplant. 1997, 19, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Pihusch, M.; Lohse, P.; Reitberger, J.; Hiller, E.; Andreesen, R.; Kolb, H.-J.; Holler, E.; Pihusch, R. Impact of thrombophilic gene mutations and graft-versus-host disease on thromboembolic complications after allogeneic hematopoietic stem-cell transplantation. Transplantation 2004, 78, 911–918. [Google Scholar] [CrossRef]
- Salat, C.; Holler, E.; Kolb, H.J.; Pihusch, R.; Reinhardt, B.; Penovici, M.; Ledderose, G.; Hiller, E. The relevance of plasminogen activator inhibitor 1 (PAI-1) as a marker for the diagnosis of hepatic veno-occlusive disease in patients after bone marrow transplantation. Leuk. Lymphoma 1999, 33, 25–32. [Google Scholar] [CrossRef]
- Salat, C.; Holler, E.; Kolb, H.J.; Reinhardt, B.; Pihusch, R.; Wilmanns, W.; Hiller, E. Plasminogen activator inhibitor-1 confirms the diagnosis of hepatic veno-occlusive disease in patients with hyperbilirubinemia after bone marrow transplantation. Blood 1997, 89, 2184–2188. [Google Scholar] [CrossRef]
- Katsuyama, K.; Shichiri, M.; Marumo, F.; Hirata, Y. NO inhibits cytokine-induced iNOS expression and NF-κB activation by interfering with phosphorylation and degradation of IκB-α. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1796–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Kasai, K.; Gross, S.S. NO suppresses while peroxynitrite sustains NF-κB: A paradigm to rationalize cytoprotective and cytotoxic actions attributed to NO. Cardiovasc. Res. 2004, 63, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Miller, M.J.; Huang, X.; Warren, J.S. Nitric oxide modulates MCP-1 expression in endothelial cells: Implications for the pathogenesis of pulmonary granulomatous vasculitis. Inflammation 2003, 27, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.R.; Zhu, Y.; Halushka, P.V.; Lincoln, T.M.; Mendelsohn, M.E. Mechanism of platelet inhibition by nitric oxide: In vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef] [Green Version]
- Du, X. A new mechanism for nitric oxide- and cGMP-mediated platelet inhibition. Blood 2007, 109, 392–393. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, J.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. TNFalpha and oxLDL reduce protein S-nitrosylation in endothelial cells. J. Biol. Chem. 2001, 276, 41383–41387. [Google Scholar] [CrossRef] [Green Version]
- Kolluru, G.K.; Sinha, S.; Majumder, S.; Muley, A.; Siamwala, J.H.; Gupta, R.; Chatterjee, S. Shear stress promotes nitric oxide production in endothelial cells by sub-cellular delocalization of eNOS: A basis for shear stress mediated angiogenesis. Nitric Oxide Biol. Chem. 2010, 22, 304–315. [Google Scholar] [CrossRef]
- Sriram, K.; Laughlin, J.G.; Rangamani, P.; Tartakovsky, D.M. Shear-Induced Nitric Oxide Production by Endothelial Cells. Biophys. J. 2016, 111, 208–221. [Google Scholar] [CrossRef] [Green Version]
- Cunha, F.Q.; Assreuy, J.; Moss, D.W.; Rees, D.; Leal, L.M.; Moncada, S.; Carrier, M.; O’Donnell, C.A.; Liew, F.Y. Differential induction of nitric oxide synthase in various organs of the mouse during endotoxaemia: Role of TNF-alpha and IL-1-beta. Immunology 1994, 81, 211–215. [Google Scholar] [PubMed]
- Hu, G.; Su, Y.; Kang, B.H.; Fan, Z.; Dong, T.; Brown, D.R.; Cheah, J.; Wittrup, K.D.; Chen, J. High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat. Commun. 2021, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Weisser, S.B.; McLarren, K.W.; Kuroda, E.; Sly, L.M. Generation and characterization of murine alternatively activated macrophages. Methods Mol. Biol. 2013, 946, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Kirkebøen, K.A.; Strand, O.A. The role of nitric oxide in sepsis--an overview. Acta Anaesthesiol. Scand. 1999, 43, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, P.; Chaudry, I.H. Endothelial nitric oxide synthase is downregulated during hyperdynamic sepsis. Biochim. Biophys. Acta 1997, 1335, 182–190. [Google Scholar] [CrossRef]
- Lima, R.; Serone, A.P.; Schor, N.; Higa, E.M. Effect of cyclosporin A on nitric oxide production in cultured LLC-PK1 cells. Ren. Fail. 2001, 23, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Sudhir, K.; MacGregor, J.S.; DeMarco, T.; De Groot, C.J.; Taylor, R.N.; Chou, T.M.; Yock, P.G.; Chatterjee, K. Cyclosporine impairs release of endothelium-derived relaxing factors in epicardial and resistance coronary arteries. Circulation 1994, 90, 3018–3023. [Google Scholar] [CrossRef] [Green Version]
- Drobyski, W.; Keever, C.; Hanson, G.; McAuliffe, T.; Griffith, O. Inhibition of nitric oxide production is associated with enhanced weight loss, decreased survival, and impaired alloengraftment in mice undergoing graft-versus-host disease after bone marrow transplantation. Blood 1994, 84, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D.; Wang, X.; Kanel, G.C.; Ito, Y.; Bethea, N.W.; McCuskey, M.K.; Tokes, Z.A.; Tsai, J.; McCuskey, R.S. Decreased hepatic nitric oxide production contributes to the development of rat sinusoidal obstruction syndrome. Hepatology 2003, 38, 900–908. [Google Scholar] [CrossRef]
- Batts, E.D.; Lazarus, H.M. Diagnosis and treatment of transplantation-associated thrombotic microangiopathy: Real progress or are we still waiting? Bone Marrow Transplant. 2007, 40, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.E.; Adamson, R.H. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loghmani, H.; Conway, E.M. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Nishida, O.; Kuriyama, N.; Nakamura, T.; Kawaji, T.; Onouchi, T.; Hasegawa, D.; Shimomura, Y. Effects of Thrombomodulin in Reducing Lethality and Suppressing Neutrophil Extracellular Trap Formation in the Lungs and Liver in a Lipopolysaccharide-Induced Murine Septic Shock Model. Int. J. Mol. Sci. 2021, 22, 4933. [Google Scholar] [CrossRef]
- Joyce, D.E.; Gelbert, L.; Ciaccia, A.; DeHoff, B.; Grinnell, B.W. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J. Biol. Chem. 2001, 276, 11199–11203. [Google Scholar] [CrossRef] [Green Version]
- Neyrinck, A.P.; Liu, K.D.; Howard, J.P.; Matthay, M.A. Protective mechanisms of activated protein C in severe inflammatory disorders. Br. J. Pharmacol. 2009, 158, 1034–1047. [Google Scholar] [CrossRef] [Green Version]
- Hirata, N.; Ngo, D.T.; Phan, P.H.; Ainai, A.; Phung, T.T.B.; Ta, T.A.; Takasaki, J.; Kawachi, S.; Nunoi, H.; Nakajima, N.; et al. Recombinant human thrombomodulin for pneumonia-induced severe ARDS complicated by DIC in children: A preliminary study. J. Anesth. 2021, 35, 638–645. [Google Scholar] [CrossRef]
- Kadono, K.; Uchida, Y.; Hirao, H.; Miyauchi, T.; Watanabe, T.; Iida, T.; Ueda, S.; Kanazawa, A.; Mori, A.; Okajima, H.; et al. Thrombomodulin Attenuates Inflammatory Damage Due to Liver Ischemia and Reperfusion Injury in Mice in Toll-Like Receptor 4-Dependent Manner. Am. J. Transplant. 2017, 17, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kunz, G.; Ohlin, A.-K.; Adami, A.; Zöller, B.; Svensson, P.; Lane, D.A. Naturally occurring mutations in the thrombomodulin gene leading to impaired expression and function. Blood 2002, 99, 3646–3653. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Konishi, A.; Tsubokura, Y.; Azuma, Y.; Hotta, M.; Yoshimura, H.; Nakanishi, T.; Fujita, S.; Satake, A.; Katayama, Y.; et al. Effects of recombinant thrombomodulin on long-term prognosis after allogeneic hematopoietic stem cell transplantation. Transpl. Immunol. 2019, 57, 101247. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Maruyama, I.; Shimazaki, S.; Yamamoto, Y.; Aikawa, N.; Ohno, R.; Hirayama, A.; Matsuda, T.; Asakura, H.; Nakashima, M.; et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: Results of a phase III, randomized, double-blind clinical trial. J. Thromb. Haemost. 2007, 5, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shirahata, A.; Mimuro, J.; Takahashi, H.; Tsuji, H.; Kitajima, I.; Matsushita, T.; Eguchi, Y.; Kitamura, N.; Honda, G.; Sakata, Y. Postmarketing Surveillance of Recombinant Human Soluble Thrombomodulin (Thrombomodulin α) in Pediatric Patients With Disseminated Intravascular Coagulation. Clin. Appl. Thromb./Hemost. 2014, 20, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Shirahata, A.; Mimuro, J.; Takahashi, H.; Kitajima, I.; Tsuji, H.; Eguchi, Y.; Matsushita, T.; Kajiki, M.; Honda, G.; Sakata, Y. Recombinant soluble human thrombomodulin (thrombomodulin alfa) in the treatment of neonatal disseminated intravascular coagulation. Eur. J. Pediatr. 2014, 173, 303–311. [Google Scholar] [CrossRef]
- Noris, M.; Mescia, F.; Remuzzi, G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat. Rev. Nephrol. 2012, 8, 622–633. [Google Scholar] [CrossRef]
- Waters, A.M.; Licht, C. aHUS caused by complement dysregulation: New therapies on the horizon. Pediatr. Nephrol. 2011, 26, 41–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Józsi, M.; Licht, C.; Strobel, S.; Zipfel, S.L.H.; Richter, H.; Heinen, S.; Zipfel, P.F.; Skerka, C. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008, 111, 1512–1514. [Google Scholar] [CrossRef] [Green Version]
- Moore, I.; Strain, L.; Pappworth, I.; Kavanagh, D.; Barlow, P.N.; Herbert, A.P.; Schmidt, C.Q.; Staniforth, S.J.; Holmes, L.V.; Ward, R.; et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010, 115, 379–387. [Google Scholar] [CrossRef]
- Delvaeye, M.; DeVriese, A.; Moons, M.; Esmon, N.; Esmon, C.; Conway, E.M. Regulation of Complement Activation by Thrombomodulin. Blood 2009, 114, 5127. [Google Scholar] [CrossRef]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Elhadad, S.; Chapin, J.; Copertino, D.; Van Besien, K.; Ahamed, J.; Laurence, J. MASP2 levels are elevated in thrombotic microangiopathies: Association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin. Exp. Immunol. 2021, 203, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Yamashita, K.; Mizugishi, K.; Watanabe, T.; Sakamoto, S.; Kitano, T.; Kondo, T.; Kawabata, H.; Kadowaki, N.; Takaori-Kondo, A. Serum neutrophil extracellular trap levels predict thrombotic microangiopathy after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2013, 19, 1683–1689. [Google Scholar] [CrossRef] [Green Version]
- Boros, M.; Szalay, L.; Kaszaki, J. Endothelin-1 induces mucosal mast cell degranulation and tissue injury via ETA receptors. Clin. Sci. 2002, 103 (Suppl. 48), 31S–34S. [Google Scholar] [CrossRef] [Green Version]
- Helset, E.; Sildnes, T.; Konopski, Z.S. Endothelin-1 Stimulates Monocytes in vitro to Release Chemotactic Activity Identified as Interleukin-8 and Monocyte Chemotactic Protein-1. Mediators Inflamm. 1994, 3, 155–160. [Google Scholar] [CrossRef] [Green Version]
- McCarron, R.M.; Wang, L.; Stanimirovic, D.B.; Spatz, M. Endothelin induction of adhesion molecule expression on human brain microvascular endothelial cells. Neurosci. Lett. 1993, 156, 31–34. [Google Scholar] [CrossRef]
- Tschaikowsky, K.; Sägner, S.; Lehnert, N.; Kaul, M.; Ritter, J. Endothelin in septic patients: Effects on cardiovascular and renal function and its relationship to proinflammatory cytokines. Crit. Care Med. 2000, 28, 1854–1860. [Google Scholar] [CrossRef]
- Danesh, A.; Inglis, H.C.; Jackman, R.P.; Wu, S.; Deng, X.; Muench, M.O.; Heitman, J.W.; Norris, P.J. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood 2014, 123, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, M.; Mitsuiki, N.; Yanagimachi, M.; Yamamoto, M.; Fukuda, T.; Miura, O.; Oba, R.; Igarashi, A.; Nagata, K.; Morio, T. Utility of novel T-cell-specific extracellular vesicles in monitoring and evaluation of acute GVHD. Int. J. Hematol. 2021, 113, 910–920. [Google Scholar] [CrossRef]
- Hildebrandt, G.C.; Chao, N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br. J. Haematol. 2020, 190, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, S.; Michonneau, D.; Sicre de Fontbrune, F.; Sutra del Galy, A.; Xhaard, A.; Robin, M.; Peffault de Latour, R.; Socie, G. Allogeneic reactivity–mediated endothelial cell complications after HSCT: A plea for consensual definitions. Blood Adv. 2019, 3, 2424–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle, J.-H.; Giralt, S.A. Hepatic Veno-Occlusive Disease after Hematopoietic Stem Cell Transplantation: Risk Factors and Stratification, Prophylaxis, and Treatment. Biol. Blood Marrow Transplant. 2016, 22, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.D.; McCuskey, R.S.; Wang, X.; Hu, L.; McCuskey, M.K.; Epstein, R.B.; Kanel, G.C. Characterization of a reproducible rat model of hepatic veno-occlusive disease. Hepatology 1999, 29, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, M.; Wei, M.; Lu, B.; Wu, X.; Wang, Z.; Ji, L. Liver Inflammatory Injury Initiated by DAMPs-TLR4-MyD88/TRIF-NFκB Signaling Pathway Is Involved in Monocrotaline-Induced HSOS. Toxicol. Sci. 2019, 172, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Zhang, Q.; Mumaw, C.L.; Raiker, N.; Yu, J.; Velez de Mendizabal, N.; Haneline, L.S.; Robertson, K.A.; Skiles, J.; Diaz-Ricart, M.; et al. Biomarkers for Diagnosis and Prognosis of Sinusoidal Obstruction Syndrome after Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1739–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondón, G.; Saliba, R.M.; Chen, J.; Ledesma, C.; Alousi, A.M.; Oran, B.; Hosing, C.M.; Kebriaei, P.; Khouri, I.F.; Shpall, E.J.; et al. Impact of Fluid Overload as New Toxicity Category on Hematopoietic Stem Cell Transplantation Outcomes. Biol. Blood Marrow Transplant. 2017, 23, 2166–2171. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-J.; Lee, K.-H.; Lee, J.-H.; Lee, J.-H.; Kim, S.; Seol, M.; Lee, Y.-S.; Kim, W.-K.; Park, C.-J.; Chi, H.-S.; et al. Peri-engraftment clinical abnormalities following allogeneic hematopoietic cell transplantation: A retrospective review of 216 patients. Bone Marrow Transplant. 2003, 32, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Varma, A.; Rondon, G.; Srour, S.A.; Chen, J.; Ledesma, C.; Champlin, R.E.; Ciurea, S.O.; Saliba, R.M. Endothelial Activation and Stress Index (EASIX) at Admission Predicts Fluid Overload in Recipients of Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 1013–1020. [Google Scholar] [CrossRef]
- Janin, A.; Deschaumes, C.; Daneshpouy, M.; Estaquier, J.; Micic-Polianski, J.; Rajagopalan-Levasseur, P.; Akarid, K.; Mounier, N.; Gluckman, E.; Socié, G.; et al. CD95 engagement induces disseminated endothelial cell apoptosis in vivo: Immunopathologic implications. Blood 2002, 99, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Gerbitz, A.; Nickoloff, B.J.; Olkiewicz, K.; Willmarth, N.E.; Hildebrandt, G.; Liu, C.; Kobzik, L.; Eissner, G.; Holler, E.; Ferrara, J.L.M.; et al. A role for tumor necrosis factor-alpha-mediated endothelial apoptosis in the development of experimental idiopathic pneumonia syndrome. Transplantation 2004, 78, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, G.C.; Olkiewicz, K.M.; Corrion, L.A.; Chang, Y.; Clouthier, S.G.; Liu, C.; Cooke, K.R. Donor-derived TNF-α regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood 2004, 104, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, T.; Slack, J.; Slatter, M.A.; O’Brien, C.; Cant, A.; Thomas, M.; Brodlie, M.; Annavarapu, S.; Gennery, A.R. Endothelial cell damage in idiopathic pneumonia syndrome. Bone Marrow Transplant. 2018, 53, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Renaud, C.; Kuypers, J.M.; Chiu, C.Y.; Huang, M.-L.; Samayoa, E.; Xie, H.; Yu, G.; Fisher, C.E.; Gooley, T.A.; et al. Idiopathic pneumonia syndrome after hematopoietic cell transplantation: Evidence of occult infectious etiologies. Blood 2015, 125, 3789–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.M.; Mammoto, T.; Schultz, A.; Yuan, H.-T.; Christiani, D.; Karumanchi, S.A.; Sukhatme, V.P. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006, 3, e46. [Google Scholar] [CrossRef] [PubMed]
- Whitney, J.E.; Feng, R.; Koterba, N.; Chen, F.; Bush, J.; Graham, K.; Lacey, S.F.; Melenhorst, J.J.; Parikh, S.M.; Weiss, S.L.; et al. Endothelial Biomarkers Are Associated With Indirect Lung Injury in Sepsis-Associated Pediatric Acute Respiratory Distress Syndrome. Crit. Care Explor. 2020, 2, e0295. [Google Scholar] [CrossRef]
- Spitzer, T.R. Engraftment syndrome: Double-edged sword of hematopoietic cell transplants. Bone Marrow Transplant. 2015, 50, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Maiolino, A.; Biasoli, I.; Lima, J.; Portugal, A.C.; Pulcheri, W.; Nucci, M. Engraftment syndrome following autologous hematopoietic stem cell transplantation: Definition of diagnostic criteria. Bone Marrow Transplant. 2003, 31, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Oyama, Y.; Cohen, B.; Traynor, A.; Brush, M.; Rodriguez, J.; Burt, R.K. Engraftment syndrome: A common cause for rash and fever following autologous hematopoietic stem cell transplantation for multiple sclerosis. Bone Marrow Transplant. 2002, 29, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Kumar, S.K.; Buadi, F.; Dingli, D.; Litzow, M.R.; Gastineau, D.A.; Inwards, D.J.; Elliott, M.A.; et al. Peripheral blood stem cell transplant for POEMS syndrome is associated with high rates of engraftment syndrome. Eur. J. Haematol. 2008, 80, 397–406. [Google Scholar] [CrossRef]
- Koreth, J.; Biernacki, M.; Aldridge, J.; Kim, H.T.; Alyea, E.P., 3rd; Armand, P.; Cutler, C.; Ho, V.T.; Wu, C.J.; Antin, J.H.; et al. Syngeneic donor hematopoietic stem cell transplantation is associated with high rates of engraftment syndrome. Biol. Blood Marrow Transplant. 2011, 17, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omer, A.K.; Kim, H.T.; Yalamarti, B.; McAfee, S.L.; Dey, B.R.; Ballen, K.K.; Attar, E.; Chen, Y.-B.; Spitzer, T.R. Engraftment syndrome after allogeneic hematopoietic cell transplantation in adults. Am. J. Hematol. 2014, 89, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Frame, D.; Braun, T.; Gatza, E.; Hanauer, D.A.; Zhao, S.; Magenau, J.M.; Schultz, K.; Tokala, H.; Ferrara, J.L.M.; et al. Engraftment syndrome after allogeneic hematopoietic cell transplantation predicts poor outcomes. Biol. Blood Marrow Transplant. 2014, 20, 1407–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruutu, T.; Barosi, G.; Benjamin, R.J.; Clark, R.E.; George, J.N.; Gratwohl, A.; Holler, E.; Iacobelli, M.; Kentouche, K.; Lämmle, B.; et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: Results of a consensus process by an International Working Group. Haematologica 2007, 92, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.T.; Cutler, C.; Carter, S.; Martin, P.; Adams, R.; Horowitz, M.; Ferrara, J.; Soiffer, R.; Giralt, S. Blood and marrow transplant clinical trials network toxicity committee consensus summary: Thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2005, 11, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Jodele, S.; Laskin, B.L.; Dandoy, C.E.; Myers, K.C.; El-Bietar, J.; Davies, S.M.; Goebel, J.; Dixon, B.P. A new paradigm: Diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2015, 29, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Tichelli, A.; Gratwohl, A. Vascular endothelium as “novel” target of graft-versus-host disease. Best Pract. Res. Clin. Haematol. 2008, 21, 139–148. [Google Scholar] [CrossRef]
- Kraft, S.; Bollinger, N.; Bodenmann, B.; Heim, D.; Bucher, C.; Lengerke, C.; Kleber, M.; Tsakiris, D.A.; Passweg, J.; Tzankov, A.; et al. High mortality in hematopoietic stem cell transplant-associated thrombotic microangiopathy with and without concomitant acute graft-versus-host disease. Bone Marrow Transplant. 2019, 54, 540–548. [Google Scholar] [CrossRef]
- Li, A.; Wu, Q.; Davis, C.; Kirtane, K.S.; Pham, P.D.; Sorror, M.L.; Lee, S.J.; Gopal, A.K.; Dong, J.-F.; Garcia, D.A.; et al. Transplant-Associated Thrombotic Microangiopathy Is a Multifactorial Disease Unresponsive to Immunosuppressant Withdrawal. Biol. Blood Marrow Transplant. 2019, 25, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Hamaguchi, M.; Hirabayashi, N.; Haneda, M.; Terakura, S.; Atsuta, Y.; Imagama, S.; Kanie, T.; Murata, M.; Taji, H.; et al. Intestinal thrombotic microangiopathy after allogeneic bone marrow transplantation: A clinical imitator of acute enteric graft-versus-host disease. Bone Marrow Transplant. 2004, 33, 1143–1150. [Google Scholar] [CrossRef]
- El-Bietar, J.; Warren, M.; Dandoy, C.; Myers, K.C.; Lane, A.; Wallace, G.; Davies, S.M.; Jodele, S. Histologic Features of Intestinal Thrombotic Microangiopathy in Pediatric and Young Adult Patients after Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1994–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, M.; Jodele, S.; Dandoy, C.; Myers, K.C.; Wallace, G.; Nelson, A.; El-Bietar, J. A Complete Histologic Approach to Gastrointestinal Biopsy From Hematopoietic Stem Cell Transplant Patients With Evidence of Transplant-Associated Gastrointestinal Thrombotic Microangiopathy. Arch. Pathol. Lab. Med. 2017, 141, 1558–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskin, B.L.; Maisel, J.; Goebel, J.; Yin, H.J.; Luo, G.; Khoury, J.C.; Davies, S.M.; Jodele, S. Renal arteriolar C4d deposition: A novel characteristic of hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transplantation 2013, 96, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jodele, S.; Davies, S.M.; Lane, A.; Khoury, J.; Dandoy, C.; Goebel, J.; Myers, K.; Grimley, M.; Bleesing, J.; El-Bietar, J.; et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: A study in children and young adults. Blood 2014, 124, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Wang, J.; Chen, J.; Su, J.; Tang, Y.; Wu, X.; Ma, X.; Chen, F.; Ruan, C.; Zheng, X.L.; et al. Plasma levels of complement activation fragments C3b and sC5b-9 significantly increased in patients with thrombotic microangiopathy after allogeneic stem cell transplantation. Ann. Hematol. 2017, 96, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Jodele, S.; Licht, C.; Goebel, J.; Dixon, B.P.; Zhang, K.; Sivakumaran, T.A.; Davies, S.M.; Pluthero, F.G.; Lu, L.; Laskin, B.L. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 2013, 122, 2003–2007. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Sakellari, I.; Chatzikonstantinou, T.; Mallouri, D.; Batsis, I.; Vardi, A.; Bousiou, Z.; Koravou, E.-E.; Masmanidou, M.; Touloumenidou, T.; et al. Endothelial and Complement Activation As Predictors of Survival in Adult Allogeneic Hematopoietic Cell Transplantation. HemaSphere 2021, 5, e487. [Google Scholar] [CrossRef]
- Jodele, S.; Dandoy, C.E.; Myers, K.C.; El-Bietar, J.; Nelson, A.; Wallace, G.; Laskin, B.L. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transfus. Apher. Sci. 2016, 54, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J. Nitric oxide in transplantation-related thrombotic microangiopathy. Bone Marrow Transplant. 2009, 43, 513–514. [Google Scholar] [CrossRef]
- Trapp, A.; Weis, M. The impact of immunosuppression on endothelial function. J. Cardiovasc. Pharmacol. 2005, 45, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Keller, T.T.; Mairuhu, A.T.A.; de Kruif, M.D.; Klein, S.K.; Gerdes, V.E.A.; ten Cate, H.; Brandjes, D.P.M.; Levi, M.; van Gorp, E.C.M. Infections and endothelial cells. Cardiovasc. Res. 2003, 60, 40–48. [Google Scholar] [CrossRef]
- Mezö, B.; Horváth, O.; Sinkovits, G.; Veszeli, N.; Kriván, G.; Prohászka, Z. Validation of Early Increase in Complement Activation Marker sC5b-9 as a Predictive Biomarker for the Development of Thrombotic Microangiopathy After Stem Cell Transplantation. Front. Med. 2020, 7, 646. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Nakamae, H.; Shindo, T.; Ohtani, K.; Hidaka, Y.; Ohtsuka, Y.; Makuuchi, Y.; Kuno, M.; Takakuwa, T.; Harada, N.; et al. Early Elevation of Complement Factor Ba Is a Predictive Biomarker for Transplant-Associated Thrombotic Microangiopathy. Front. Immunol. 2021, 12, 2669. [Google Scholar] [CrossRef] [PubMed]
- Horváth, O.; Kállay, K.; Csuka, D.; Mező, B.; Sinkovits, G.; Kassa, C.; Stréhn, A.; Csordás, K.; Sinkó, J.; Prohászka, Z.; et al. Early Increase in Complement Terminal Pathway Activation Marker sC5b-9 Is Predictive for the Development of Thrombotic Microangiopathy after Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Mir, E.; Palomo, M.; Rovira, M.; Pereira, A.; Escolar, G.; Penack, O.; Holler, E.; Carreras, E.; Diaz-Ricart, M. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017, 52, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Luft, T.; Dietrich, S.; Falk, C.; Conzelmann, M.; Hess, M.; Benner, A.; Neumann, F.; Isermann, B.; Hegenbart, U.; Ho, A.D.; et al. Steroid-refractory GVHD: T-cell attack within a vulnerable endothelial system. Blood 2011, 118, 1685–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, S.; Falk, C.S.; Benner, A.; Karamustafa, S.; Hahn, E.; Andrulis, M.; Hegenbart, U.; Ho, A.D.; Dreger, P.; Luft, T. Endothelial Vulnerability and Endothelial Damage Are Associated with Risk of Graft-versus-Host Disease and Response to Steroid Treatment. Biol. Blood Marrow Transplant. 2013, 19, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Kernan, N.A.; Grupp, S.; Smith, A.R.; Arai, S.; Triplett, B.; Antin, J.H.; Lehmann, L.; Shore, T.; Ho, V.T.; Bunin, N.; et al. Final results from a defibrotide treatment-IND study for patients with hepatic veno-occlusive disease/sinusoidal obstruction syndrome. Br. J. Haematol. 2018, 181, 816–827. [Google Scholar] [CrossRef]
- Richardson, P.; Aggarwal, S.; Topaloglu, O.; Villa, K.F.; Corbacioglu, S. Systematic review of defibrotide studies in the treatment of veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS). Bone Marrow Transplant. 2019, 54, 1951–1962. [Google Scholar] [CrossRef] [Green Version]
- Pescador, R.; Capuzzi, L.; Mantovani, M.; Fulgenzi, A.; Ferrero, M.E. Defibrotide: Properties and clinical use of an old/new drug. Vascul. Pharmacol. 2013, 59, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Devadas, S.K.; Toshniwal, M.; Bagal, B.; Khattry, N. Successful Treatment of Transplant Associated Thrombotic Microangiopathy (TA-TMA) with Low Dose Defibrotide. Indian J. Hematol. Blood Transfus. 2018, 34, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Soiffer, R.J.; Antin, J.H.; Uno, H.; Jin, Z.; Kurtzberg, J.; Martin, P.L.; Steinbach, G.; Murray, K.F.; Vogelsang, G.B.; et al. Defibrotide for the Treatment of Severe Hepatic Veno-Occlusive Disease and Multiorgan Failure after Stem Cell Transplantation: A Multicenter, Randomized, Dose-Finding Trial. Biol. Blood Marrow Transplant. 2010, 16, 1005–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Bernal, D.; Palomo, M.; Martínez, C.M.; Millán-Rivero, J.E.; García-Guillén, A.I.; Blanquer, M.; Díaz-Ricart, M.; Sackstein, R.; Carreras, E.; Moraleda, J.M. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J. Cell. Mol. Med. 2020, 24, 8031–8044. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J. Hematopoietic cell transplantation-associated thrombotic microangiopathy: A review of pathophysiology, diagnosis, and treatment. J. Blood Med. 2016, 7, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Jodele, S.; Fukuda, T.; Vinks, A.; Mizuno, K.; Laskin, B.L.; Goebel, J.; Dixon, B.P.; Teusink, A.; Pluthero, F.G.; Lu, L.; et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol. Blood Marrow Transplant. 2014, 20, 518–525. [Google Scholar] [CrossRef] [Green Version]
- De Fontbrune, F.S.; Galambrun, C.; Sirvent, A.; Huynh, A.; Faguer, S.; Nguyen, S.; Bay, J.-O.; Neven, B.; Moussi, J.; Simon, L.; et al. Use of Eculizumab in Patients With Allogeneic Stem Cell Transplant-Associated Thrombotic Microangiopathy: A Study From the SFGM-TC. Transplantation 2015, 99, 1953–1959. [Google Scholar] [CrossRef]
- Jan, A.S.; Hosing, C.; Aung, F.; Yeh, J. Approaching treatment of transplant-associated thrombotic Microangiopathy from two directions with Eculizumab and transitioning from Tacrolimus to Sirolimus. Transfusion 2019, 59, 3519–3524. [Google Scholar] [CrossRef]
- Zhang, R.; Zhou, M.; Qi, J.; Miao, W.; Zhang, Z.; Wu, D.; Han, Y. Efficacy and Safety of Eculizumab in the Treatment of Transplant-Associated Thrombotic Microangiopathy: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 11, 3486. [Google Scholar] [CrossRef]
- Rambaldi, A.; Gritti, G.; Micò, M.C.; Frigeni, M.; Borleri, G.; Salvi, A.; Landi, F.; Pavoni, C.; Sonzogni, A.; Gianatti, A.; et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology 2020, 225, 152001. [Google Scholar] [CrossRef]
- Poppelaars, F.; Faria, B.; Schwaeble, W.; Daha, M.R. The contribution of complement to the pathogenesis of IgA nephropathy: Are complement-targeted therapies moving from rare disorders to more common diseases? J. Clin. Med. 2021, 10, 4715. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, J.-X.; Xu, X.-F.; Zhang, X.-X.; Ye, X.-N.; Huang, J. Successful treatment of plasma exchange-refractory thrombotic thrombocytopenic purpura with rituximab: A case report. World J. Clin. Cases 2020, 8, 2617–2622. [Google Scholar] [CrossRef] [PubMed]
- Niewold, T.B.; Alpert, D.; Scanzello, C.R.; Paget, S.A. Rituximab treatment of thrombotic thrombocytopenic purpura in the setting of connective tissue disease. J. Rheumatol. 2006, 33, 1194–1196. [Google Scholar] [PubMed]
- Marr, H.; McDonald, E.-J.; Merriman, E.; Smith, M.; Mangos, H.; Stoddart, C.; Ganly, P. Successful treatment of transplant-associated microangiopathy with rituximab. N. Z. Med. J. 2009, 122, 72–74. [Google Scholar] [PubMed]
- Ostronoff, M.; Ostronoff, F.; Calixto, R.; Florêncio, R.; Florêncio, M.; Domingues, M.C.; Souto Maior, A.P.; Sucupira, A.; Tagliari, C. Life-threatening hemolytic-uremic syndrome treated with rituximab in an allogeneic bone marrow transplant recipient. Bone Marrow Transplant. 2007, 39, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Gallerani, E.; Lerch, E.; Romagnani, E.; Stathis, A.; Giardelli, G.; Zwhalen, H.; Marone, C.; Cavalli, F. Thrombotic thrombocytopenic purpura associated with renal failure after autologous transplantation for multiple myeloma successfully treated with rituximab. Eur. J. Haematol. 2006, 77, 527–529. [Google Scholar] [CrossRef]
- Rosenthal, R.A.; Chukwuogo, N.A.; Ocasio, V.H.; Kahng, K.U. Cyclosporine inhibits endothelial cell prostacyclin production. J. Surg. Res. 1989, 46, 593–596. [Google Scholar] [CrossRef]
- Voss, B.L.; Hamilton, K.K.; Samara, E.N.; McKee, P.A. Cyclosporine suppression of endothelial prostacyclin generation. A possible mechanism for nephrotoxicity. Transplantation 1988, 45, 793–796. [Google Scholar] [CrossRef]
- Conde, M.; Andrade, J.; Bedoya, F.J.; Santa Maria, C.; Sobrino, F. Inhibitory effect of cyclosporin A and FK506 on nitric oxide production by cultured macrophages. Evidence of a direct effect on nitric oxide synthase activity. Immunology 1995, 84, 476–481. [Google Scholar]
- Hernández, G.L.; Volpert, O.V.; Iñiguez, M.A.; Lorenzo, E.; Martínez-Martínez, S.; Grau, R.; Fresno, M.; Redondo, J.M. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: Roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med. 2001, 193, 607–620. [Google Scholar] [CrossRef]
- Wolff, D.; Wilhelm, S.; Hahn, J.; Gentilini, C.; Hilgendorf, I.; Steiner, B.; Kahl, C.; Junghanss, C.; Hartung, G.; Casper, J.; et al. Replacement of calcineurin inhibitors with daclizumab in patients with transplantation-associated microangiopathy or renal insufficiency associated with graft-versus-host disease. Bone Marrow Transplant. 2006, 38, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Laskin, B.L.; Goebel, J.; Davies, S.M.; Jodele, S. Small vessels, big trouble in the kidneys and beyond: Hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood 2011, 118, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jodele, S.; Laskin, B.L.; Goebel, J.; Khoury, J.C.; Pinkard, S.L.; Carey, P.M.; Davies, S.M. Does early initiation of therapeutic plasma exchange improve outcome in pediatric stem cell transplant-associated thrombotic microangiopathy? Transfusion 2013, 53, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.S.; Xenocostas, A.; Lipton, J.H. Transplantation-associated thrombotic microangiopathy: Twenty-two years later. Bone Marrow Transplant. 2002, 30, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Sartain, S.; Shubert, S.; Wu, M.-F.; Srivaths, P.; Teruya, J.; Krance, R.; Martinez, C. Therapeutic Plasma Exchange does not Improve Renal Function in Hematopoietic Stem Cell Transplantation-Associated Thrombotic Microangiopathy: An Institutional Experience. Biol. Blood Marrow Transplant. 2019, 25, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, M.A.; Vesely, S.K.; George, J.N.; Chandler, L.; Duvall, D.; Smith, J.W.; Gilcher, R.O. Complications of plasma exchange in 71 consecutive patients treated for clinically suspected thrombotic thrombocytopenic purpura-hemolytic-uremic syndrome. Transfusion 2000, 40, 896–901. [Google Scholar] [CrossRef]
- Nguyen, L.; Terrell, D.R.; Duvall, D.; Vesely, S.K.; George, J.N. Complications of plasma exchange in patients treated for thrombotic thrombocytopenic purpura. IV. An additional study of 43 consecutive patients, 2005 to 2008. Transfusion 2009, 49, 392–394. [Google Scholar] [CrossRef]
- Howard, M.A.; Williams, L.A.; Terrell, D.R.; Duvall, D.; Vesely, S.K.; George, J.N. Complications of plasma exchange in patients treated for clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Transfusion 2006, 46, 154–156. [Google Scholar] [CrossRef]
- Yamamoto, S.; Toyama, D.; Sugishita, Y.; Kaneko, R.; Okamoto, N.; Koganesawa, M.; Fujita, S.; Akiyama, K.; Matsuno, R.; Isoyama, K. Prophylactic recombinant thrombomodulin treatment prevents hepatic sinusoidal obstruction syndrome in high-risk pediatric patients that undergo hematopoietic stem cell transplants. Pediatr. Transplant. 2018, 22, e13269. [Google Scholar] [CrossRef]
- Sakai, M.; Ikezoe, T.; Bandobashi, K.; Togitani, K.; Yokoyama, A. Successful treatment of transplantation-associated thrombotic microangiopathy with recombinant human soluble thrombomodulin. Bone Marrow Transplant. 2010, 45, 803–805. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Akamatsu, N.; Ichida, A.; Kaneko, J.; Arita, J.; Hasegawa, K.; Sakamoto, Y.; Kokudo, N. Possible efficacy of recombinant human soluble thrombomodulin for the treatment of thrombotic microangiopathy after liver transplantation. Liver Transplant. 2016, 22, 689–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Maeda, Y.; Sando, Y.; Nakamura, M.; Tani, K.; Ishikawa, T.; Nishimori, H.; Matsuoka, K.-I.; Fujii, N.; Kondo, E.; et al. Treatment of thrombotic microangiopathy after hematopoietic stem cell transplantation with recombinant human soluble thrombomodulin. Transfusion 2016, 56, 886–892. [Google Scholar] [CrossRef]
- Otsuka, Y.; Kondo, T.; Nomura, R.; Yamashita, K.; Takaori-Kondo, A. Successful treatment with recombinant thrombomodulin in transplant-associated thrombotic microangiopathy following HLA-haploidentical transplantation. Rinsho. Ketsueki. 2019, 60, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Takeuchi, A.; Taniguchi, A.; Togitani, K.; Yokoyama, A. Recombinant human soluble thrombomodulin counteracts capillary leakage associated with engraftment syndrome. Bone Marrow Transplant. 2011, 46, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Togitani, K.; Komatsu, N.; Isaka, M.; Yokoyama, A. Successful treatment of sinusoidal obstructive syndrome after hematopoietic stem cell transplantation with recombinant human soluble thrombomodulin. Bone Marrow Transplant. 2010, 45, 783–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, J.; Kurauchi, K.; Fukano, R.; Noguchi, M.; Okamura, J. Heterogeneous response to recombinant thrombomodulin by grade of sinusoidal obstructive syndrome after pediatric stem cell transplantation. Bone Marrow Transplant. 2016, 51, 1543–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, F.; Forster, A.; Chenevard, R.; Enseleit, F.; Hürlimann, D.; Corti, R.; Spieker, L.E.; Frey, D.; Hermann, M.; Riesen, W.; et al. Simvastatin improves endothelial function in patients with rheumatoid arthritis. J. Am. Coll. Cardiol. 2005, 45, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Reriani, M.K.; Dunlay, S.M.; Gupta, B.; West, C.P.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Effects of statins on coronary and peripheral endothelial function in humans: A systematic review and meta-analysis of randomized controlled trials. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 704–716. [Google Scholar] [CrossRef]
- Ii, M.; Losordo, D.W. Statins and the endothelium. Vascul. Pharmacol. 2007, 46, 1–9. [Google Scholar] [CrossRef]
- Zheng, P.; Wu, Q.-L.; Li, B.-B.; Chen, P.; Nie, D.-M.; Zhang, R.; Fang, J.; Xia, L.-H.; Hong, M. Simvastatin ameliorates graft-vs-host disease by regulating angiopoietin-1 and angiopoietin-2 in a murine model. Leuk. Res. 2017, 55, 49–54. [Google Scholar] [CrossRef]
- Jiang, S.; Penack, O.; Terzer, T.; Schult, D.; Majer-Lauterbach, J.; Radujkovic, A.; Blau, I.W.; Bullinger, L.; Müller-Tidow, C.; Dreger, P.; et al. Predicting sinusoidal obstruction syndrome after allogeneic stem cell transplantation with the EASIX biomarker panel. Haematologica 2021, 106, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutbier, B.; Jiang, X.; Dietert, K.; Ehrler, C.; Lienau, J.; Van Slyke, P.; Kim, H.; Hoang, V.C.; Maynes, J.T.; Dumont, D.J.; et al. Vasculotide reduces pulmonary hyperpermeability in experimental pneumococcal pneumonia. Crit. Care 2017, 21, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latreille, E.; Lee, W.L. Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy. Viruses 2021, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Yan, T.; Chopp, M.; Zacharek, A.; Ning, R.; Van Slyke, P.; Dumont, D.; Landschoot-Ward, J.; Liang, L.; Chen, J. Angiopoietin-1 Mimetic Peptide Promotes Neuroprotection after Stroke in Type 1 Diabetic Rats. Cell Transplant. 2018, 27, 1744–1752. [Google Scholar] [CrossRef] [Green Version]
- Venkat, P.; Ning, R.; Zacharek, A.; Culmone, L.; Liang, L.; Landschoot-Ward, J.; Chopp, M. Treatment with an Angiopoietin-1 mimetic peptide promotes neurological recovery after stroke in diabetic rats. CNS Neurosci. Ther. 2021, 27, 48–59. [Google Scholar] [CrossRef]
- Trieu, M.; van Meurs, M.; van Leeuwen, A.L.I.; Van Slyke, P.; Hoang, V.; Geeraedts, L.M.G.J.; Boer, C.; van den Brom, C.E. Vasculotide, an Angiopoietin-1 Mimetic, Restores Microcirculatory Perfusion and Microvascular Leakage and Decreases Fluid Resuscitation Requirements in Hemorrhagic Shock. Anesthesiology 2018, 128, 361–374. [Google Scholar] [CrossRef]
- Sanwal, R.; Joshi, K.; Ditmans, M.; Tsai, S.S.H.; Lee, W.L. Ultrasound and Microbubbles for Targeted Drug Delivery to the Lung Endothelium in ARDS: Cellular Mechanisms and Therapeutic Opportunities. Biomedicines 2021, 9, 803. [Google Scholar] [CrossRef]
- Subramaniyam, D.; Virtala, R.; Pawłowski, K.; Clausen, I.G.; Warkentin, S.; Stevens, T.; Janciauskiene, S. TNF-alpha-induced self expression in human lung endothelial cells is inhibited by native and oxidized alpha1-antitrypsin. Int. J. Biochem. Cell Biol. 2008, 40, 258–271. [Google Scholar] [CrossRef]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a Target for Therapeutic Interventions. Front. Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, L.; Morin, F.; Robin, M.; Peyneau, M.; Schlageter, M.H.; Desmier, D.; Pagliuca, S.; Sutra Del Galy, A.; Sicre de Fontbrune, F.; Xhaard, A.; et al. Human-Derived α1-Antitrypsin is Still Efficacious in Heavily Pretreated Patients with Steroid-Resistant Gastrointestinal Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 2020, 26, 1620–1626. [Google Scholar] [CrossRef]
- Ostrovsky, O.; Shimoni, A.; Baryakh, P.; Morgulis, Y.; Mayorov, M.; Beider, K.; Shteingauz, A.; Ilan, N.; Vlodavsky, I.; Nagler, A. Modification of heparanase gene expression in response to conditioning and LPS treatment: Strong correlation to rs4693608 SNP. J. Leukoc. Biol. 2014, 95, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taflin, C.; Favier, B.; Baudhuin, J.; Savenay, A.; Hemon, P.; Bensussan, A.; Charron, D.; Glotz, D.; Mooney, N. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. USA 2011, 108, 2891–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenswil, K.J.G.; Pisterzi, P.; Sánchez-Duffhues, G.; van Dijk, C.; Lolli, A.; Knuth, C.; Vanchin, B.; Jaramillo, A.C.; Hoogenboezem, R.M.; Sanders, M.A.; et al. Endothelium-derived stromal cells contribute to hematopoietic bone marrow niche formation. Cell Stem Cell 2021, 28, 653–670.e11. [Google Scholar] [CrossRef]
- Kopp, H.-G.; Avecilla, S.T.; Hooper, A.T.; Rafii, S. The bone marrow vascular niche: Home of HSC differentiation and mobilization. Physiology 2005, 20, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milone, G.; Bellofiore, C.; Leotta, S.; Milone, G.A.; Cupri, A.; Duminuco, A.; Garibaldi, B.; Palumbo, G. Endothelial Dysfunction after Hematopoietic Stem Cell Transplantation: A Review Based on Physiopathology. J. Clin. Med. 2022, 11, 623. https://doi.org/10.3390/jcm11030623
Milone G, Bellofiore C, Leotta S, Milone GA, Cupri A, Duminuco A, Garibaldi B, Palumbo G. Endothelial Dysfunction after Hematopoietic Stem Cell Transplantation: A Review Based on Physiopathology. Journal of Clinical Medicine. 2022; 11(3):623. https://doi.org/10.3390/jcm11030623
Chicago/Turabian StyleMilone, Giuseppe, Claudia Bellofiore, Salvatore Leotta, Giulio Antonio Milone, Alessandra Cupri, Andrea Duminuco, Bruno Garibaldi, and Giuseppe Palumbo. 2022. "Endothelial Dysfunction after Hematopoietic Stem Cell Transplantation: A Review Based on Physiopathology" Journal of Clinical Medicine 11, no. 3: 623. https://doi.org/10.3390/jcm11030623
APA StyleMilone, G., Bellofiore, C., Leotta, S., Milone, G. A., Cupri, A., Duminuco, A., Garibaldi, B., & Palumbo, G. (2022). Endothelial Dysfunction after Hematopoietic Stem Cell Transplantation: A Review Based on Physiopathology. Journal of Clinical Medicine, 11(3), 623. https://doi.org/10.3390/jcm11030623