
Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience

 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Diagnosis of ML II—Biochemical and Molecular Genetic Features
3.2. Biochemical Features of Hyperparathyroidism
3.3. Radiological Abnormalities Identified
3.4. Management and Clinical Course
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heo, J.S.; Choi, K.Y.; Sohn, S.H.; Kim, C.; Kim, Y.J.; Shin, S.H.; Lee, J.M.; Lee, J.; Sohn, J.A.; Lim, B.C.; et al. A case of mucolipidosis II presenting with prenatal skeletal dysplasia and severe secondary hyperparathyroidism at birth. Korean J. Pediatr. 2012, 55, 438–444. [Google Scholar] [CrossRef]
- Yokoi, A.; Niida, Y.; Kuroda, M.; Imi-Hashida, Y.; Toma, T.; Yachie, A. B-cell-specific accumulation of inclusion bodies loaded with HLA class II molecules in patients with mucolipidosis II (I-cell disease). Pediatr. Res. 2018, 86, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.G.; DeMars, R.I. Mutant Enzymatic and Cytological Phenotypes in Cultured Human Fibroblasts. Science 1967, 157, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.W.; Wiedemann, H.-R. The genetic mucolipidoses. Qual. Life Res. 1970, 9, 113–139. [Google Scholar] [CrossRef]
- Velho, R.V.; Harms, F.L.; Danyukova, T.; Ludwig, N.F.; Friez, M.J.; Cathey, S.S.; Filocamo, M.; Tappino, B.; Gunes, N.; Tuysuz, B.; et al. The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations. Hum. Mutat. 2019, 40, 842–864. [Google Scholar] [PubMed]
- Sly, W.; Sundaram, V. The I-Cell model: The molecular basis for abnormal lysosomal enzyme transport in mucolipidosis II and mucolipidosis III. In Genetic and Metabolic Disease in Pediatrics; Lloyd, K.K., Scriver, C.R., Eds.; Butterworths: London, UK, 1985; pp. 91–110. [Google Scholar]
- Ammer, L.S.; Oussoren, E.; Muschol, N.M.; Pohl, S.; Rubio-Gozalbo, M.E.; Santer, R.; Stuecker, R.; Vettorazzi, E.; Breyer, S.R. Hip Morphology in Mucolipidosis Type II. J. Clin. Med. 2020, 9, 728. [Google Scholar] [CrossRef]
- Leyva, C.; Buch, M.; Wierenga, K.J.; Berkovitz, G.; Seeherunvong, T. A neonate with mucolipidosis II and transient secondary hyperparathyroidism. J. Pediatr. Endocrinol. Metab. 2019, 32, 1399–1402. [Google Scholar] [CrossRef]
- David-Vizcarra, G.; Briody, J.; Ault, J.; Fietz, M.; Fletcher, J.; Savarirayan, R.; Wilson, M.; McGill, J.; Edwards, M.; Munns, C.; et al. The natural history and osteodystrophy of mucolipidosis types II and III. J. Paediatr. Child Health 2010, 46, 316–322. [Google Scholar] [CrossRef]
- Sathasivam, A.; Garibaldi, L.; Murphy, R.; Ibrahim, J. Transient Neonatal Hyperparathyroidism: A Presenting Feature of Mucolipidosis Type II. J. Pediatr. Endocrinol. Metab. 2006, 19, 859–862. [Google Scholar] [CrossRef]
- Alfadhel, M.; Alshehhi, W.; Alshaalan, H.; Al Balwi, M.; Eyaid, W. Mucolipidosis II: First report from Saudi Arabia. Ann. Saudi Med. 2013, 33, 382–386, Erratum in Ann. Saudi Med. 2014, 34, 91. [Google Scholar] [CrossRef]
- Khan, A.; Ho, J.; Pender, A.; Wei, X.; Potter, M. I-Cell Disease (Mucolipidosis II) Presenting as Neonatal Fractures: A Case for Continued Monitoring of Serum Parathyroid Hormone Levels. Clin. Pediatr. Endocrinol. 2008, 17, 81–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, M.H.-C.; Pitukcheewanont, P. Mucolipidosis type II (I-cell disease) masquerading as rickets: Two case reports and review of literature. J. Pediatr. Endocrinol. Metab. 2012, 25, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Alegra, T.; Cury, G.; Todeschini, L.A.; Schwartz, I.V. Should neonatal hyperparathyroidism associated with mucolipidosis II/III be treated pharmacologically? J. Pediatr. Endocrinol. Metab. 2013, 26, 1011–1013. [Google Scholar] [CrossRef]
- Pazzaglia, U.E.; Beluffi, G.; Campbell, J.B.; Bianchi, E.; Colavita, N.; Diard, F.; Gugliantini, P.; Hirche, U.; Kozlowski, K.; Marchi, A.; et al. Mucolipidosis II: Correlation between radiological features and histopathology of the bones. Pediatr. Radiol. 1989, 19, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, U.E.; Beluffi, G.; Castello, A.; Coci, A.; Zatti, G. Bone changes of mucolipidosis II at different ages. Postmortem study of three cases. Clin. Orthop. Relat. Res. 1992, 276, 283–290. [Google Scholar] [CrossRef]
- Pazzaglia, U.E.; Beluffi, G.; Danesino, C.; Frediani, P.V.; Pagani, G.; Zatti, G. Neonatal mucolipidosis 2. The spontaneous evolution of early bone lesions and the effect of vitamin D treatment. Pediatr. Radiol. 1989, 20, 80–84. [Google Scholar] [CrossRef]
- Kornfeld, S.; Sly, W. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.H., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill, Inc.: New York, NY, USA, 2001; pp. 3469–3482. [Google Scholar]
- Alegra, T.; Koppe, T.; Acosta, A.; Sarno, M.; Burin, M.; Kessler, R.G.; Sperb-Ludwig, F.; Cury, G.; Baldo, G.; Matte, U.; et al. Pitfalls in the prenatal diagnosis of mucolipidosis II alpha/beta: A case report. Meta Gene 2014, 2, 403–406. [Google Scholar] [CrossRef]
- Ma, G.-C.; Ke, Y.-Y.; Chang, S.-P.; Lee, D.-J.; Chen, M. A compound heterozygous GNPTAB mutation causes mucolipidosis II with marked hair color change in a Han Chinese baby. Am. J. Med. Genet. Part A 2011, 155, 931–934. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, L.D.S.; Girisha, K.M.; Satyamoorthy, K.; Lacerda, L.; Prata, M.J.; Alves, S. Mucolipidosis type II α/β with a homozygous missense mutation in the GNPTAB gene. Am. J. Med. Genet. Part A 2012, 158A, 1225–1228. [Google Scholar] [CrossRef]
- Pinto, R.R.; Caseiro, C.; Lemos, M.; Lopes, L.; Fontes, A.; Ribeiro, H.; Pinto, E.; Silva, E.; Rocha, S.; Marcão, A.; et al. Prevalence of lysosomal storage diseases in Portugal. Eur. J. Hum. Genet. 2003, 12, 87–92. [Google Scholar] [CrossRef]
- Okada, S.; Owada, M.; Sakiyama, T.; Yutaka, T.; Ogawa, M. I-cell disease: Clinical studies of 21 Japanese cases. Clin. Genet. 2008, 28, 207–215. [Google Scholar] [CrossRef]
- Poorthuis, B.; Wevers, R.; Kleijer, W.; Groener, J.; De Jong, J.; Van Weely, S.; Niezen-Koning, K.; Van Diggelen, O. The frequency of lysosomal storage diseases in The Netherlands. Qual. Life Res. 1999, 105, 151–156. [Google Scholar] [CrossRef]
- Kovacevic, A.; Schranz, D.; Meissner, T.; Pillekamp, F.; Schmidt, K.G. Mucolipidosis II complicated by severe pulmonary hypertension. Mol. Genet. Metab. 2011, 104, 192–193. [Google Scholar] [CrossRef] [PubMed]
- McElligott, F.; Beatty, E.; O’Sullivan, S.; Hughes, J.; Lambert, D.; Cooper, A.; Crushell, E. Incidence of I-cell disease (muco-lipidosis type II) in the irish population. J. Inherit. Metab. Dis. 2011, 34, S206. [Google Scholar]
- Unger, S.; Paul, D.A.; Nino, M.C.; McKay, C.P.; Miller, S.; Sochett, E.; Braverman, N.; Clarke, J.T.R.; Cole, D.E.C.; Superti-Furga, A. Mucolipidosis II presenting as severe neonatal hyperparathyroidism. Eur. J. Pediatr. 2004, 164, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Babcock, D.S.; Bove, K.E.; Hug, G.; Dignan, P.S.J.; Soukup, S.; Warren, N.S. Fetal mucolipidosis II (I-cell disease): Radiologic and pathologic correlation. Pediatr. Radiol. 1986, 16, 32–39. [Google Scholar] [CrossRef]
- Kollmann, K.; Pestka, J.M.; Kühn, S.C.; Schöne, E.; Schweizer, M.; Karkmann, K.; Otomo, T.; Catala-Lehnen, P.; Failla, A.V.; Marshall, R.P.; et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol. Med. 2013, 5, 1871–1886. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | PTH Reference Range: 11–25 ng/L | Ca Reference Range: 2.15–2.65 mmol/L | P Reference Range: 1.2–2.0 mmol/L | ALP Reference Range: 60–550 IU/L | Vitamin D Reference Range: >50 nmol/L | Radiological Features | Treatment | |
|---|---|---|---|---|---|---|---|---|
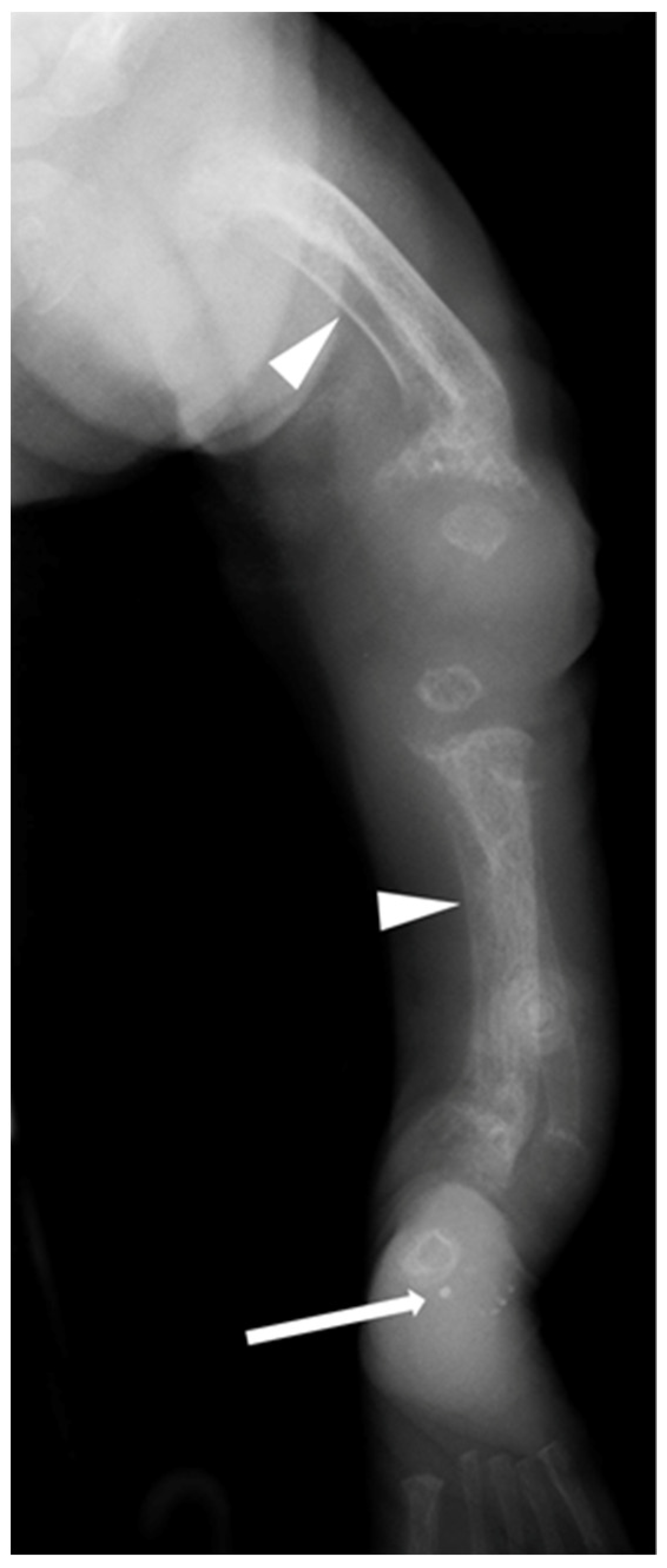
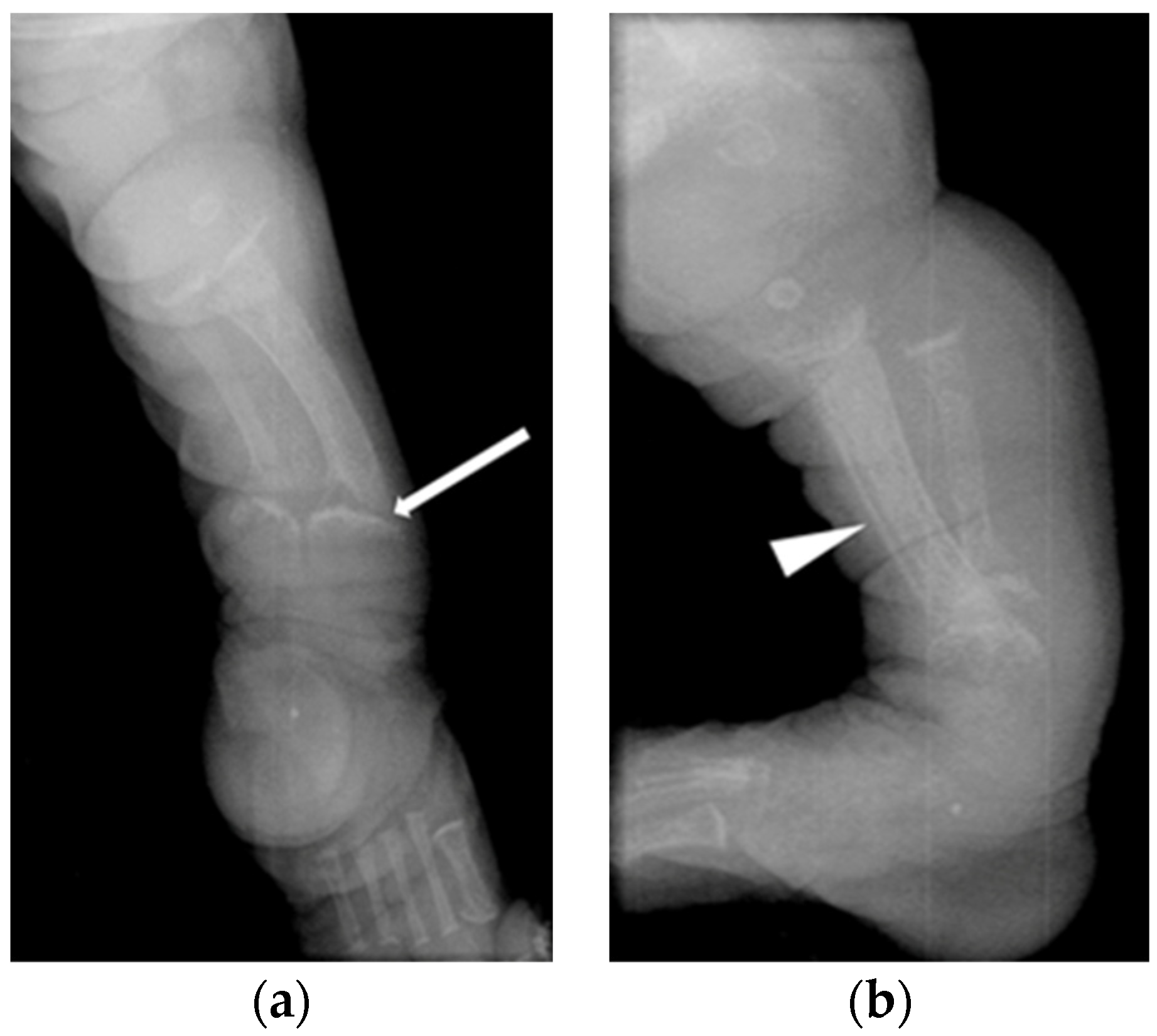
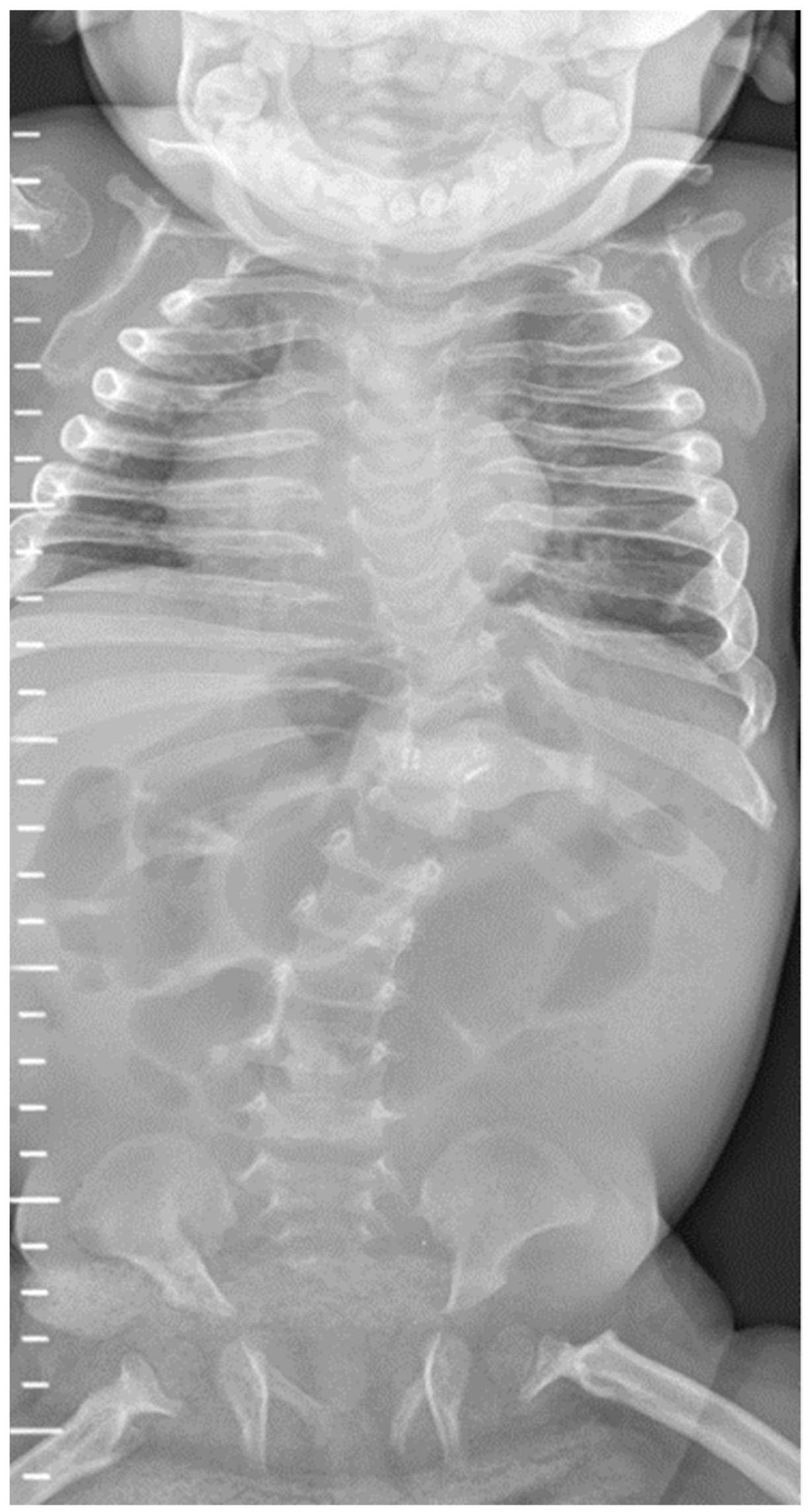
| Patient 1 | Antenatal fractures of long bones | 25 | 2.46 | 1.65 | 434 | Not available in medical notes | All patients had features of HPT (Figure 1 and Figure 2), including:
| Vitamin D 600 IU, minimal handling until 1 year of age |
| Patient 2 | Antenatal fractures of long bones | 119 ↑ | 2.36 | 1.17 | 1540 ↑ | 66.7 | None | |
| Patient 3 | No fractures | 72 ↑ | 2.45 | 1.96 | 1063 ↑ | 48 (reference range > 15 mmol/L) | None | |
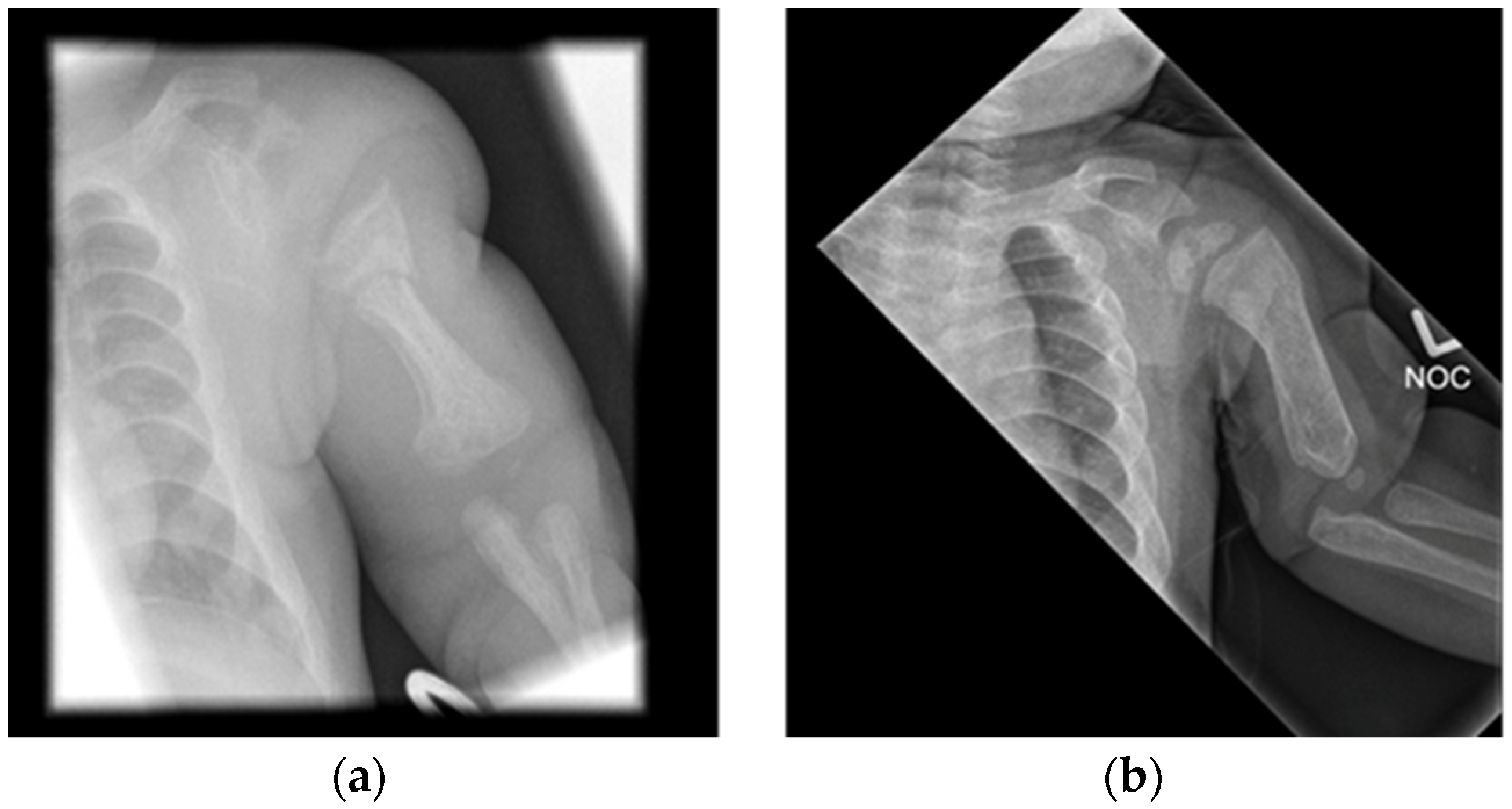
| Patient 4 | Acute fracture of the proximal left humeral neck (Figure 3a), antenatal fractures | 252 ↑ | 2.32 | 1.62 | 1007 ↑ | 121 | Vitamin D 600 IU, minimal handling for 5 month | |
| Patient 5 | No fractures | 110 ↑ | 2.31 | 1.3 | 1256 ↑ | 30 ↓ | Vitamin D 1000 IU, re-evaluation planned in due course |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boruah, R.; Monavari, A.A.; Conlon, T.; Murphy, N.; Stroiescu, A.; Ryan, S.; Hughes, J.; Knerr, I.; McDonnell, C.; Crushell, E. Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience. J. Clin. Med. 2022, 11, 1366. https://doi.org/10.3390/jcm11051366
Boruah R, Monavari AA, Conlon T, Murphy N, Stroiescu A, Ryan S, Hughes J, Knerr I, McDonnell C, Crushell E. Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience. Journal of Clinical Medicine. 2022; 11(5):1366. https://doi.org/10.3390/jcm11051366
Chicago/Turabian StyleBoruah, Ritma, Ahmad Ardeshir Monavari, Tracey Conlon, Nuala Murphy, Andreea Stroiescu, Stephanie Ryan, Joanne Hughes, Ina Knerr, Ciara McDonnell, and Ellen Crushell. 2022. "Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience" Journal of Clinical Medicine 11, no. 5: 1366. https://doi.org/10.3390/jcm11051366
APA StyleBoruah, R., Monavari, A. A., Conlon, T., Murphy, N., Stroiescu, A., Ryan, S., Hughes, J., Knerr, I., McDonnell, C., & Crushell, E. (2022). Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience. Journal of Clinical Medicine, 11(5), 1366. https://doi.org/10.3390/jcm11051366