
Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Immunohistochemistry and Genomics
3. Results
3.1. Patients
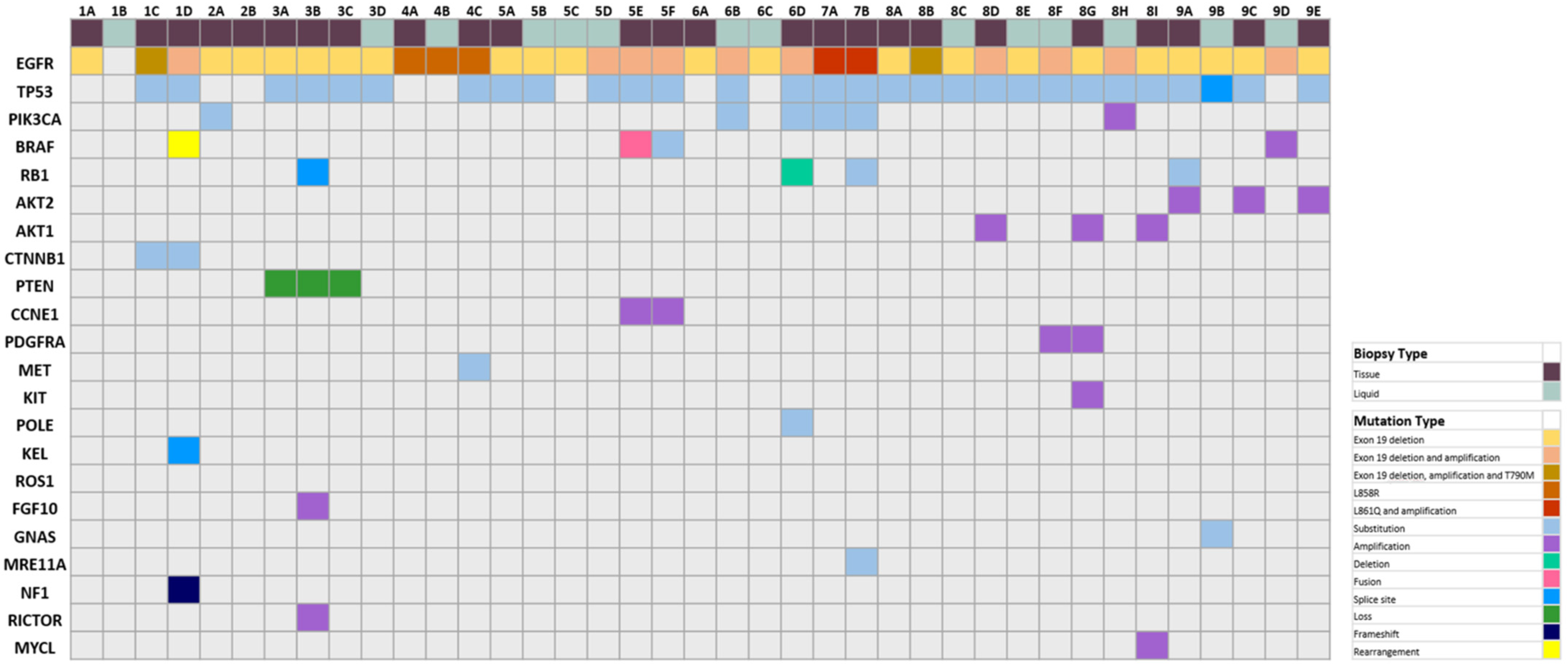
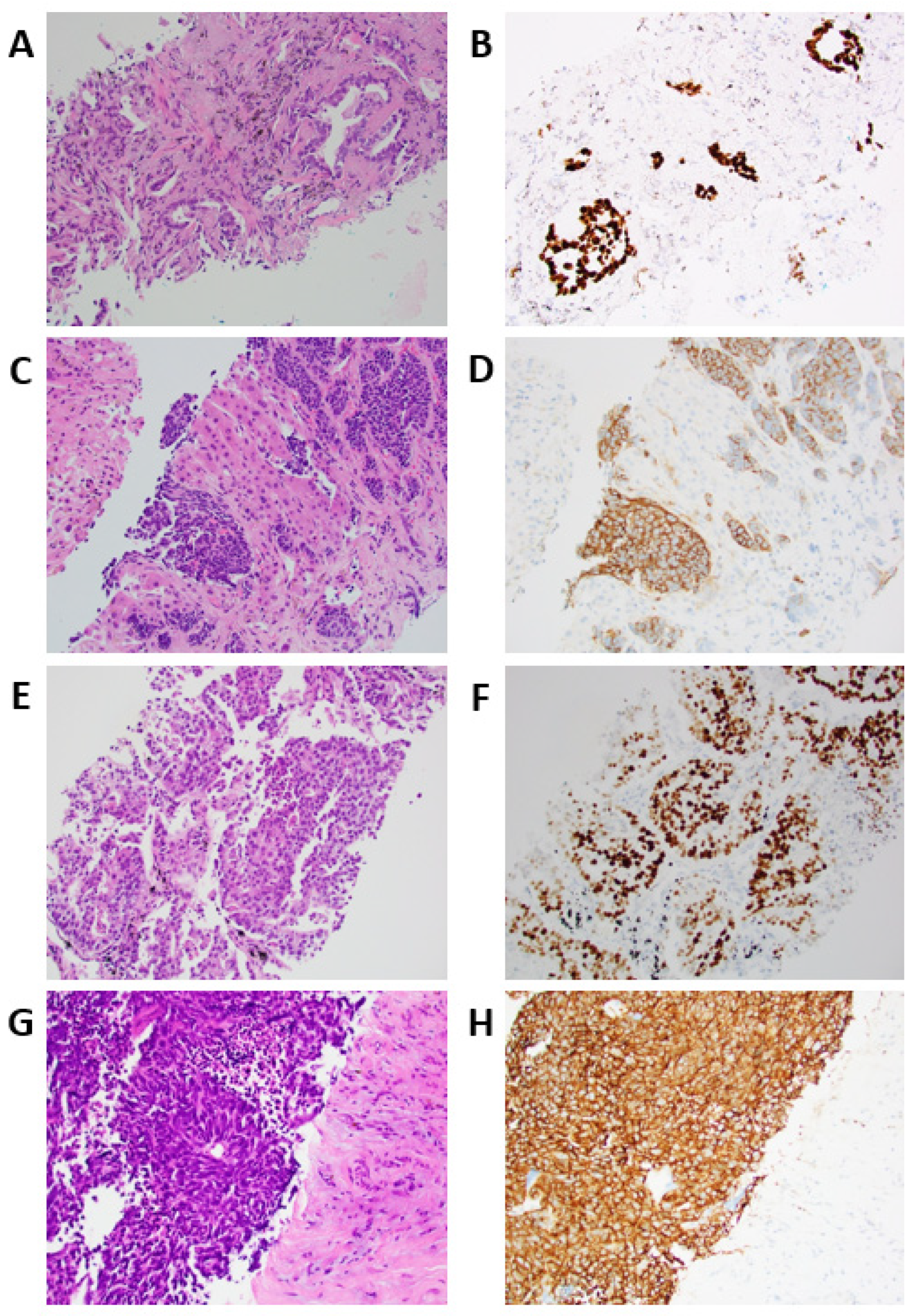
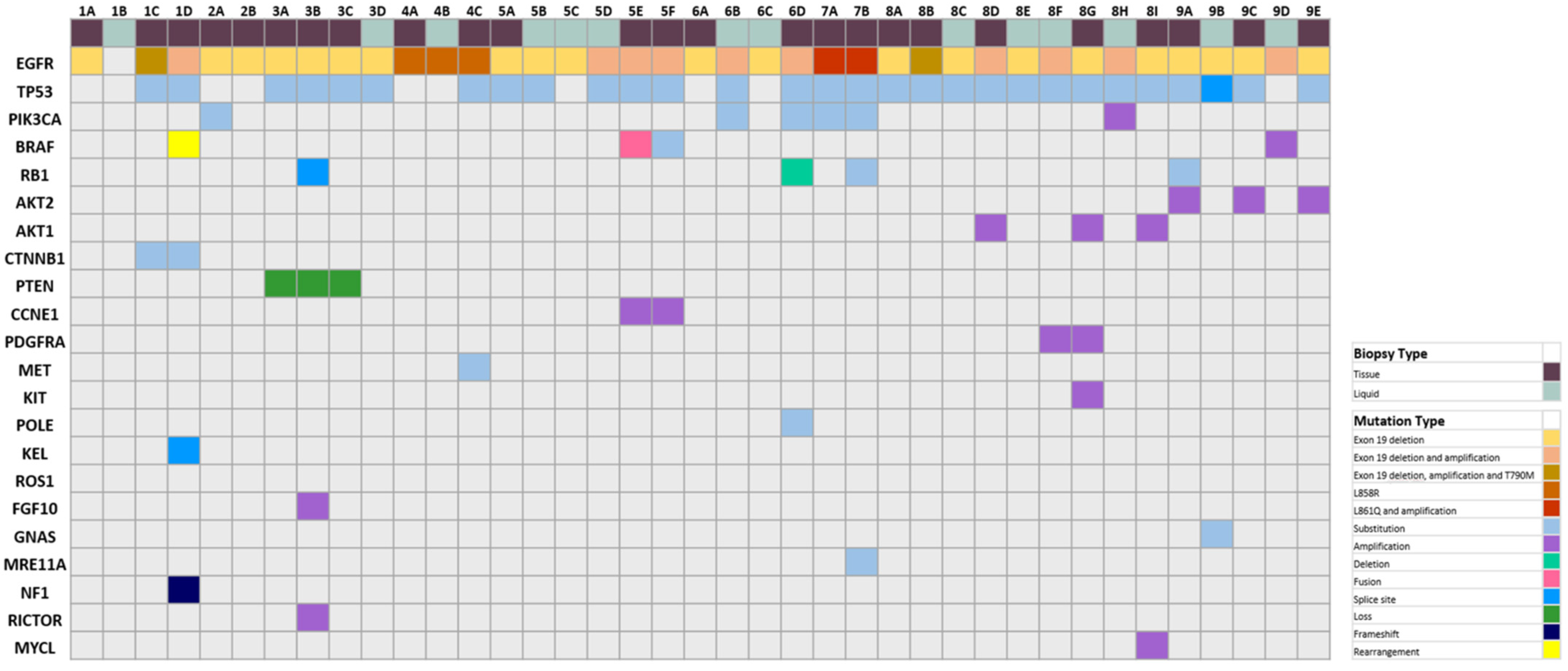
3.2. Histological and Genomic Findings
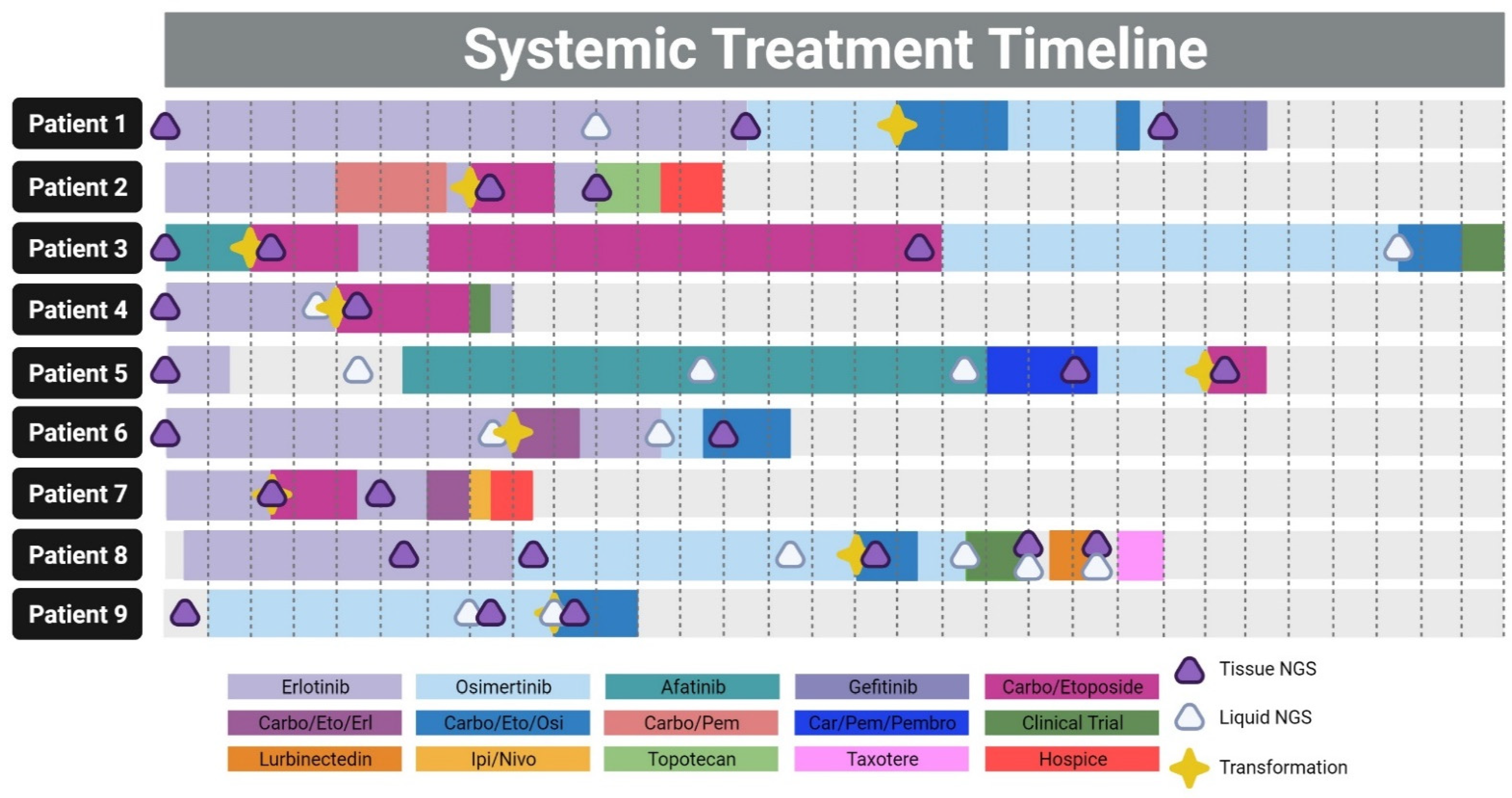
3.3. Patient Timelines and Therapies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.F.; Ren, S.X.; Li, W.; Gao, G.H. Frequency of the acquired resistant mutation T790 M in non-small cell lung cancer patients with active exon 19Del and exon 21 L858R: A systematic review and meta-analysis. BMC Cancer 2018, 18, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, T.; Akamatsu, H.; Delmonte, A.; Su, W.C.; Lee, J.S.; Chang, G.C.; Huang, X.; Jenkins, S.; Wu, Y.L. EGFR mutation analysis for prospective patient selection in AURA3 phase III trial of osimertinib versus platinum-pemetrexed in patients with EGFR T790M-positive advanced non-small-cell lung cancer. Lung Cancer 2018, 126, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Langenbucher, A.; Gupta, P.; Yoda, S.; Fetter, I.J.; Rooney, M.; Do, A.; Kem, M.; Chang, K.P.; Oh, A.Y.; et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis. Oncol. 2020, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; Villaflor, V.M. Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications. Cancers 2021, 13, 4641. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Popat, S. Histologically Transformed SCLC From EGFR-Mutant NSCLC: Understanding the Wolf in Sheep’s Clothing. J. Thorac. Oncol. 2019, 14, 1689–1691. [Google Scholar] [CrossRef]
- Song, K.A.; Niederst, M.J.; Lochmann, T.L.; Hata, A.N.; Kitai, H.; Ham, J.; Floros, K.V.; Hicks, M.A.; Hu, H.; Mulvey, H.E.; et al. Epithelial-to-Mesenchymal Transition Antagonizes Response to Targeted Therapies in Lung Cancer by Suppressing BIM. Clin. Cancer Res. 2018, 24, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Roca, E.; Gurizzan, C.; Amoroso, V.; Vermi, W.; Ferrari, V.; Berruti, A. Outcome of patients with lung adenocarcinoma with transformation to small-cell lung cancer following tyrosine kinase inhibitors treatment: A systematic review and pooled analysis. Cancer Treat. Rev. 2017, 59, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Kalemkerian, G.P. Small Cell Lung Cancer. Semin. Respir. Crit. Care Med. 2016, 37, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Pazdur, R. “Dangling” Accelerated Approvals in Oncology. N. Engl. J. Med. 2021, 384, e68. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e347. [Google Scholar] [CrossRef]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef]
- Khunger, M.; Hernandez, A.V.; Pasupuleti, V.; Rakshit, S.; Pennell, N.A.; Stevenson, J.; Mukhopadhyay, S.; Schalper, K.; Velcheti, V. Programmed Cell Death 1 (PD-1) Ligand (PD-L1) Expression in Solid Tumors As a Predictive Biomarker of Benefit From PD-1/PD-L1 Axis Inhibitors: A Systematic Review and Meta-Analysis. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.J.; Lopez-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jager, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naive Patients With Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Birer, S.R.; Dvorkin, M.; Shruti, J.; Byers, L. New Therapies and Biomarkers: Are We Ready for Personalized Treatment in Small Cell Lung Cancer? Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e276–e285. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Aldea, M.; Besse, B.; Planchard, D.; Reck, M.; Giaccone, G.; Soria, J.C. Small cell lung cancer: A slightly less orphan disease after immunotherapy. Ann. Oncol. 2021, 32, 698–709. [Google Scholar] [CrossRef]
- Ferrer, L.; Giaj Levra, M.; Brevet, M.; Antoine, M.; Mazieres, J.; Rossi, G.; Chiari, R.; Westeel, V.; Poudenx, M.; Letreut, J.; et al. A Brief Report of Transformation From NSCLC to SCLC: Molecular and Therapeutic Characteristics. J. Thorac. Oncol. 2019, 14, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Cardona, A.F.; Rojas, L.; Zatarain-Barron, Z.L.; Ruiz-Patino, A.; Ricaurte, L.; Corrales, L.; Martin, C.; Freitas, H.; Cordeiro de Lima, V.C.; Rodriguez, J.; et al. Multigene Mutation Profiling and Clinical Characteristics of Small-Cell Lung Cancer in Never-Smokers vs. Heavy Smokers (Geno1.3-CLICaP). Front. Oncol. 2019, 9, 254. [Google Scholar] [CrossRef]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, C.; Chen, H.; Jia, J.; Wang, L.; Feng, H.; Wang, H.; Song, Z.; Yang, N.; Zhang, Y. Genomic alterations and clinical outcomes in patients with lung adenocarcinoma with transformation to small cell lung cancer after treatment with EGFR tyrosine kinase inhibitors: A multicenter retrospective study. Lung Cancer 2021, 155, 20–27. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Taniguchi, H.; Zhan, Y.A.; Hasan, M.M.; Chavan, S.S.; Meng, F.; Uddin, F.; Manoj, P.; Donoghue, M.T.A.; Won, H.H.; et al. Multi-omic analysis of lung tumors defines pathways activated in neuroendocrine transformation. Cancer Discov. 2021, 11, 3028–3047. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Stewart, E.L.; Martins-Filho, S.N.; Cabanero, M.; Wang, A.; Bao, H.; Wu, X.; Patel, D.; Chen, Z.; Law, J.H.; et al. Early Detection of Multiple Resistance Mechanisms by ctDNA Profiling in a Patient With EGFR-mutant Lung Adenocarcinoma Treated With Osimertinib. Clin. Lung Cancer 2020, 21, e488–e492. [Google Scholar] [CrossRef] [PubMed]
- Boysen Fynboe Ebert, E.; McCulloch, T.; Holmskov Hansen, K.; Linnet, H.; Sorensen, B.; Meldgaard, P. Clearing of circulating tumour DNA predicts clinical response to osimertinib in EGFR mutated lung cancer patients. Lung Cancer 2020, 143, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to SCLC after Treatment with the ALK Inhibitor Alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, J.; Ofek, E.; Barshack, I.; Gottfried, T.; Zadok, O.; Kamer, I.; Urban, D.; Perelman, M.; Onn, A. Transformation to small cell lung cancer as a mechanism of resistance to immunotherapy in non-small cell lung cancer. Lung Cancer 2019, 138, 109–115. [Google Scholar] [CrossRef]
- Rath, B.; Plangger, A.; Hamilton, G. Non-small cell lung cancer-small cell lung cancer transformation as mechanism of resistance to tyrosine kinase inhibitors in lung cancer. Cancer Drug Resist. 2020, 3, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Hwang, S.H.; Han, J.; Choi, Y.-L.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J.; Park, W.-Y. Transformation to Small Cell Lung Cancer of Pulmonary Adenocarcinoma: Clinicopathologic Analysis of Six Cases. J. Pathol. Transl. Med. 2016, 50, 258–263. [Google Scholar] [CrossRef]
- Hastings, K.; Yu, H.A.; Wei, W.; Sanchez-Vega, F.; DeVeaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Chen, J.-J.; Xing, R.; Zeng, Y.-C. Combination therapy: Future directions of immunotherapy in small cell lung cancer. Transl. Oncol. 2021, 14, 100889. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Behrendt, C.E.; Slavin, T.P.; Gray, S.W.; Castillo, D.K.; Koczywas, M.; Cristea, M.C.; Babski, K.M.; Stearns, D.; Marcum, C.A.; et al. Germline mutations and age at onset of lung adenocarcinoma. Cancer 2021, 127, 2801–2806. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Antibody | Vendor | Clone | Dilution |
|---|---|---|---|
| AE1/AE3 | Leica | AE1/AE3 | Pre-diluted |
| Chromogranin | Ventana | LK2H10 | Pre-diluted |
| CDX-2 | Ventana | ERP2764Y | Pre-diluted |
| CK7 | Cell Marque | OV-TL12/30 | Pre-diluted |
| CK20 | Ventana | RAB-MONO | Pre-diluted |
| CK5/6 | Ventana | DS-16B4 | Pre-diluted |
| INSM1 | Santa Cruz | A-8 | Pre-diluted |
| p53 | Ventana | BP53-11 | Pre-diluted |
| p63 | Ventana | 4A4 | Pre-diluted |
| p40 | Ventana | BC-28 | Pre-diluted |
| Synaptophysin | Ventana | SP11 | Pre-diluted |
| Cam5.2 | Ventana | Cam5.2 | Pre-diluted |
| CD56 | Leica | CD564 | Pre-diluted |
| TTF-1 | Ventana | SP-141 | Pre-diluted |
| Demographic | Total |
|---|---|
| Median age at diagnosis (range) | 60 (35–72) |
| Median months to transformation (range) | 16 (4–49) |
| Sex no. (%) | |
| Male | 4 (44.4%) |
| Female | 5 (55.6%) |
| Race no. (%) | |
| Caucasian | 5 (55.6%) |
| Asian | 4 (44.4%) |
| Smoking status | |
| Smoker | 4 (44.4%) |
| Never-smoker | 5 (55.6%) |
| Histology at diagnosis | |
| Adenocarcinoma | 9 (100%) |
| Initial EGFR mutation | |
| Exon 19 deletion | 7 (77.8%) |
| Exon 21 (L858R) | 1 (11.1%) |
| L861Q | 1 (11.1%) |
| Therapies received prior to transformation | |
| Erlotinib | 7 |
| Osimertinib | 4 |
| Afatinib | 2 |
| Carboplatin/pemetrexed | 1 |
| Carboplatin/pemetrexed/pembrolizumab | 1 |
| Therapies received after transformation | |
| Carboplatin/etoposide | 5 |
| Carboplatin/etoposide/osimertinib | 4 |
| Erlotinib | 4 |
| Osimertinib | 3 |
| Clinical Trial | 3 |
| Carboplatin/etoposide/erlotinib | 1 |
| Gefitinib | 1 |
| Ipilimumab/nivolumab | 1 |
| Lurbinectedin | 1 |
| Taxotere | 1 |
| Topotecan | 1 |
| Histology at Diagnosis | Histologic Grade at Diagnosis | Histologic Subtype at Diagnosis | Positive Immunostains at Diagnosis | Negative Immunostains at Diagnosis | Months to Transformation | Histology at Transformation | Positive Immunostatins at Transformation | Negative Immunostains at Transformation | Site of transformation Biopsy | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Adenocarcinoma | 2 | Acinar | Keratin 7, Napsin, TTF1 | Chromogranin, CK20, Synaptophysin | 35 | small cell carcinoma | synaptophysin, chromogranin, AE1/AE1, Keratin (Oscar), | CK7, CK20, P63, P40, TTF1, Napsin A, CDX2 | Left lung |
| 2 | Adenocarcinoma | 3 | Solid | Keratin 7, TTF1 | CK20, | 16 | small cell carcinoma | AE1/AE3, CK7, TTF1, Synaptophysin | Chromogranin, P40 | Left back mass (metastatic) |
| 3 | Adenocarcinoma | 3 | Solid | TTF1, CK7 | CK20, CDX2 | 4 | small cell carcinoma | CK7, CAM5.2, AE1/AE3, TTF1, Synaptophysin, chromogranin, CD56 | CK20 | Lymph node, right anterior pericardic (metastatic) |
| 4 | Adenocarcinoma | 2 | Acinar | TTF1 | 9 | small cell carcinoma | MCK, CK7, Synaptophysin, TTF1 | Chromogranin | Left retroperitoneal node (metastatic) | |
| 5 | Adenocarcinoma | 2 | Acinar | TTF1, Napsin A, CK7, CK20 | 49 | small cell carcinoma | synaptophysin, chromogranin, TTF1, INSM1, p53, CK7 | p40, CK5/6, Napsin A, CK20 | Liver (metastatic) | |
| 6 | Adenocarcinoma | N/A | N/A | CK7, TTF1 | CK20 | 16 | small cell carcinoma | AE1/AE3, synaptophysin, chromogranin, TTF1 | Right lung | |
| 7 | Adenocarcinoma | 2 | Acinar | N/A | N/A | 6 | small cell carcinoma | Keratin 7, keratin 20, CDX2, TTF1, synaptophysin, chromogranin | Napsin A, P40 | Liver (metastatic) |
| 8 | Adenocarcinoma | 3 | Solid | TTF1 | P63 | 32 | small cell carcinoma | CK7, Synaptophysin, INSM1, TTF1, CDX2, P53 | Napsin- A, P40 | Supraclavicular, right internal mammary, pericardial phrenic lymph node (metastatic) |
| 9 | Adenocarcinoma | 2 | Acinar | TTF1, CK7, Napsin A | P40 | 18 | small cell carcinoma | CK7, Synaptophysin, chromogranin, CD56, INSM1, TTF1, NapsinA, PanCK | CK20, p63 | Liver (metastatic) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mambetsariev, I.; Arvanitis, L.; Fricke, J.; Pharaon, R.; Baroz, A.R.; Afkhami, M.; Koczywas, M.; Massarelli, E.; Salgia, R. Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. J. Clin. Med. 2022, 11, 1429. https://doi.org/10.3390/jcm11051429
Mambetsariev I, Arvanitis L, Fricke J, Pharaon R, Baroz AR, Afkhami M, Koczywas M, Massarelli E, Salgia R. Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Journal of Clinical Medicine. 2022; 11(5):1429. https://doi.org/10.3390/jcm11051429
Chicago/Turabian StyleMambetsariev, Isa, Leonidas Arvanitis, Jeremy Fricke, Rebecca Pharaon, Angel R. Baroz, Michelle Afkhami, Marianna Koczywas, Erminia Massarelli, and Ravi Salgia. 2022. "Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer" Journal of Clinical Medicine 11, no. 5: 1429. https://doi.org/10.3390/jcm11051429
APA StyleMambetsariev, I., Arvanitis, L., Fricke, J., Pharaon, R., Baroz, A. R., Afkhami, M., Koczywas, M., Massarelli, E., & Salgia, R. (2022). Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Journal of Clinical Medicine, 11(5), 1429. https://doi.org/10.3390/jcm11051429