
Scale-Up of Academic Mesenchymal Stromal Cell Production
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. GMP-Compliant Isolation and Expansion of MSCs
2.2. Continuous Production (CP)
2.3. Discontinuous Production (DP)
2.4. Environmental Monitoring
2.5. MSC Quality Controls
2.5.1. Infectious Markers
2.5.2. Microbiological Controls
2.6. MSC Characterization According to the ISCT Guidelines [24]
Cell Count and Immunophenotype Analysis
2.7. Clonogenicity Assays
2.8. Mixed Lymphocyte Reaction (MLR)
2.9. Mesodermic Differentiation
2.10. Karyotype and Telomerase Activity
2.11. Senescence Assay
2.12. Statistics
3. Results
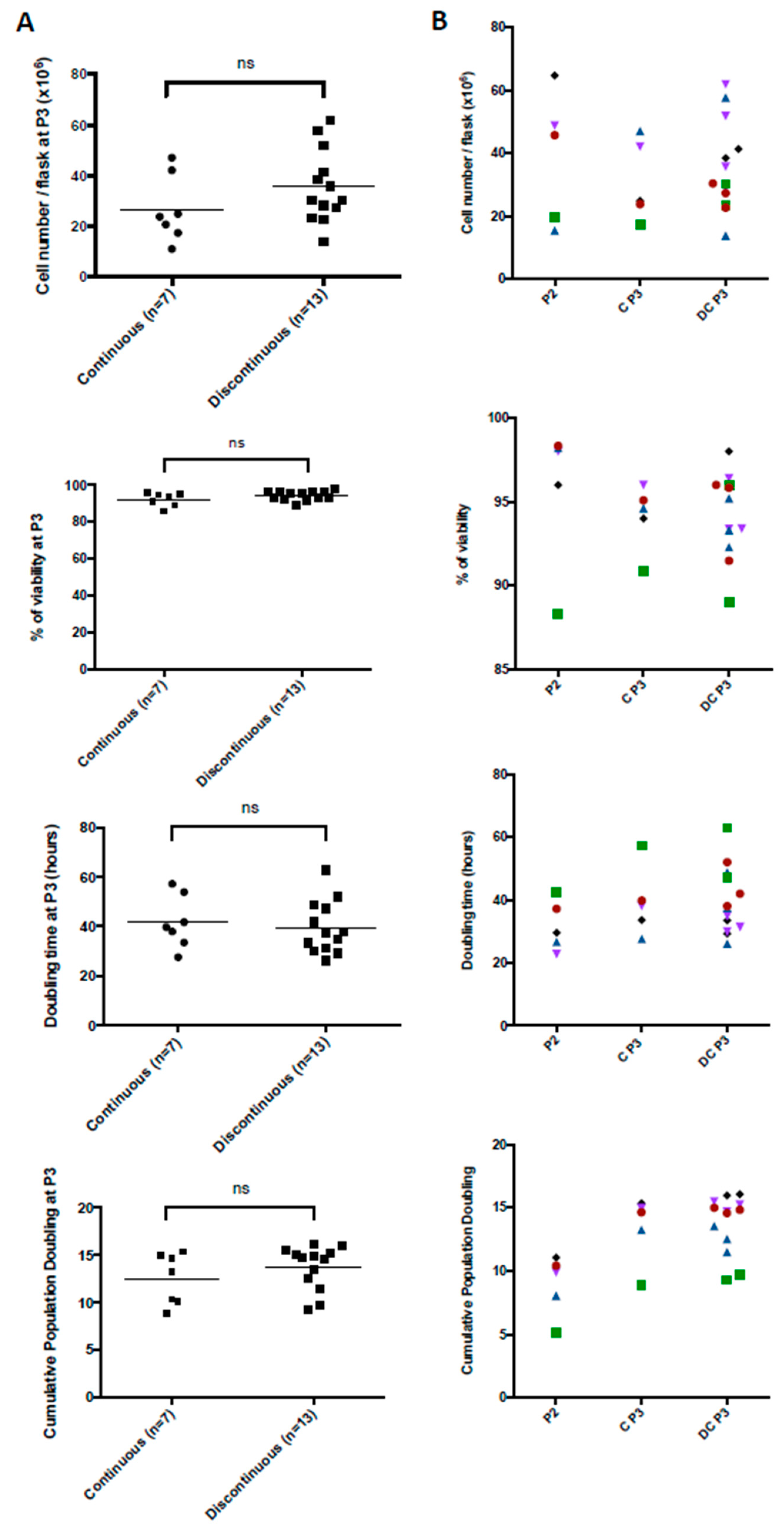
3.1. Production Data from the MSC Manufacturing Process
3.2. Particle Controls in the Production Environment
3.3. Quality Control Data from the MSC Manufacturing Process
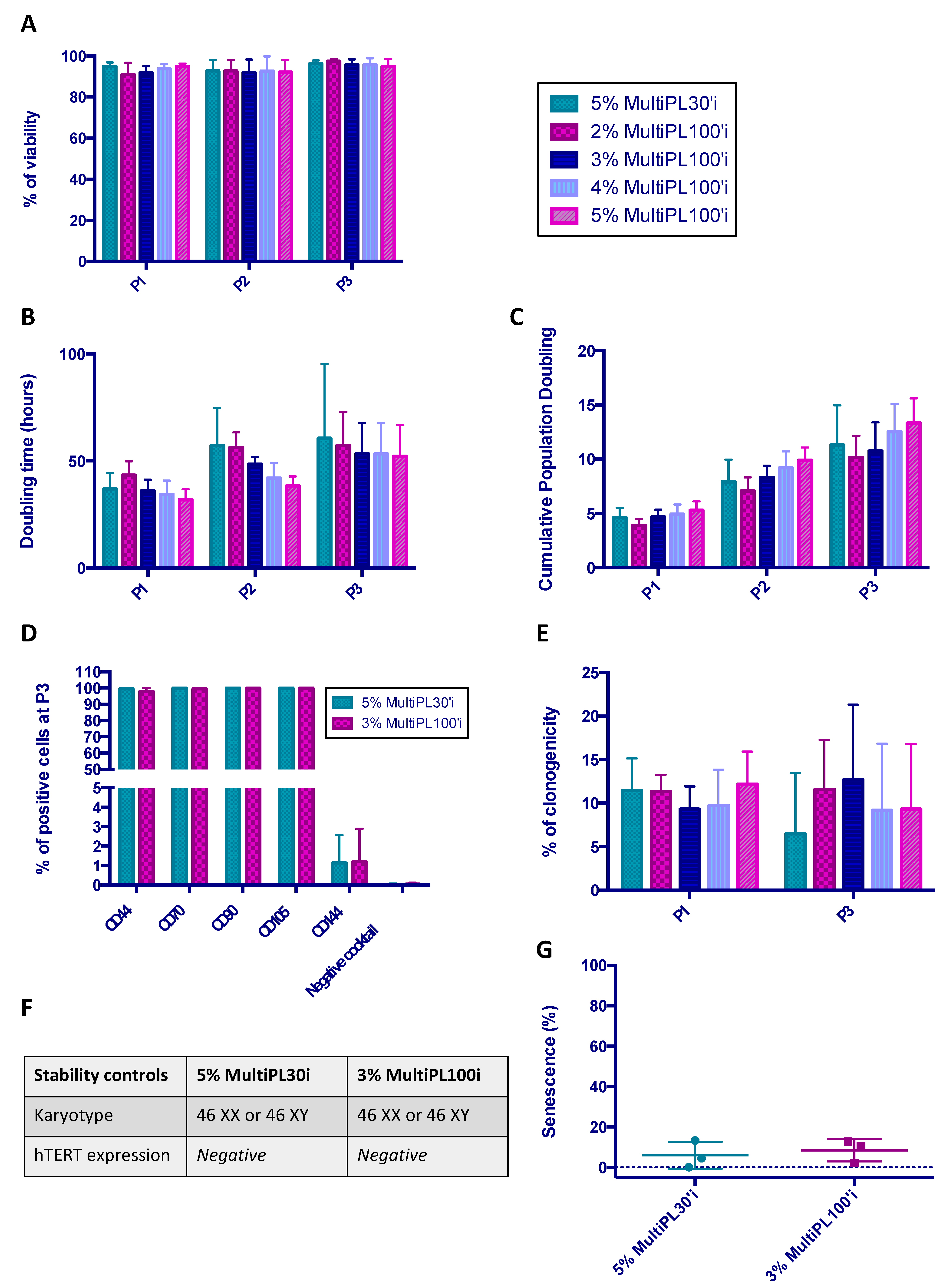
3.4. Comparison between MultiPL30i and MultiPL100i
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, M.; Mei, S.H.J.; Wolfe, D.; Champagne, J.; Fergusson, D.; Stewart, D.J.; Sullivan, K.J.; Doxtator, E.; Lalu, M.; English, S.W.; et al. Cell Therapy with Intravascular Administration of Mesenchymal Stromal Cells Continues to Appear Safe: An Updated Systematic Review and Meta-Analysis. EClinicalMedicine 2020, 19, 100249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroye, C.; Lemarié, J.; Boufenzer, A.; Labroca, P.; Cunat, L.; Alauzet, C.; Groubatch, F.; Cailac, C.; Jolly, L.; Bensoussan, D.; et al. Clinical-Grade Mesenchymal Stem Cells Derived from Umbilical Cord Improve Septic Shock in Pigs. Intensive Care Med. Exp. 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroye, C.; Gibot, S.; Reppel, L.; Bensoussan, D. Mesenchymal Stromal/Stem Cells: A New Treatment for Sepsis and Septic Shock?: MSCs in the Treatment for Sepsis and Septic Shock. Stem Cells 2017, 35, 2331–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condor, J.M.; Rodrigues, C.E.; Sousa Moreira, R.d.; Canale, D.; Volpini, R.A.; Shimizu, M.H.M.; Camara, N.O.S.; Noronha, I.D.L.; Andrade, L. Treatment with Human Whartons Jelly-Derived Mesenchymal Stem Cells Attenuates Sepsis-Induced Kidney Injury, Liver Injury, and Endothelial Dysfunction. Stem Cells Transl. Med. 2016, 5, 1048–1057. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hu, C.; Chen, L.; Tang, L.; Zhu, Y.; Xu, X.; Chen, L.; Gao, H.; Lu, X.; Yu, L.; et al. Clinical Study of Mesenchymal Stem Cell Treatment for Acute Respiratory Distress Syndrome Induced by Epidemic Influenza A (H7N9) Infection: A Hint for COVID-19 Treatment. Engineering 2020, 6, 1153–1161. [Google Scholar] [CrossRef]
- Khatri, M.; Richardson, L.A.; Meulia, T. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Influenza Virus-Induced Acute Lung Injury in a Pig Model. Stem Cell Res. Ther. 2018, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Ge, Q.; Zhang, H.; Hou, J.; Wan, L.; Cheng, W.; Wang, X.; Dong, D.; Chen, C.; Xia, J.; Guo, J.; et al. VEGF Secreted by Mesenchymal Stem Cells Mediates the Differentiation of Endothelial Progenitor Cells into Endothelial Cells via Paracrine Mechanisms. Mol. Med. Rep. 2018, 17, 1667–1675. [Google Scholar] [CrossRef] [Green Version]
- Bernard, O.; Jeny, F.; Uzunhan, Y.; Dondi, E.; Terfous, R.; Label, R.; Sutton, A.; Larghero, J.; Vanneaux, V.; Nunes, H.; et al. Mesenchymal Stem Cells Reduce Hypoxia-Induced Apoptosis in Alveolar Epithelial Cells by Modulating HIF and ROS Hypoxic Signaling. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L360–L371. [Google Scholar] [CrossRef] [Green Version]
- Loy, H.; Kuok, D.I.T.; Hui, K.P.Y.; Choi, M.H.L.; Yuen, W.; Nicholls, J.M.; Peiris, J.S.M.; Chan, M.C.W. Therapeutic Implications of Human Umbilical Cord Mesenchymal Stromal Cells in Attenuating Influenza A(H5N1) Virus–Associated Acute Lung Injury. J. Infect. Dis. 2019, 219, 186–196. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes (TNT) Is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the in Vitro and in Vivo Models of ARDS: Mitochondrial Transfer from MSC to Macrophages. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [Green Version]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in Mesenchymal Stromal Cells Induces In Vivo Recipient-Mediated Immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pochon, C.; Notarantonio, A.; Laroye, C.; Reppel, L.; Bensoussan, D.; Bertrand, A.; Rubio, M.; D’Aveni, M. Wharton’s Jelly-derived Stromal Cells and Their Cell Therapy Applications in Allogeneic Haematopoietic Stem Cell Transplantation. J. Cell. Mol. Med. 2022, 26, 1339–1350. [Google Scholar] [CrossRef]
- Liang, B.; Chen, J.; Li, T.; Wu, H.; Yang, W.; Li, Y.; Yu, C.; Nie, F.; Ma, Z.; Yang, M.; et al. Clinical Remission of a Critically Ill COVID-19 Patient Treated by Human Umbilical Cord Mesenchymal Stem Cells. Medicine 2020, 99, e21429. [Google Scholar] [CrossRef] [PubMed]
- Leng, Z.; Zhu, R.; Hou, W.; Feng, Y.; Yang, Y.; Han, Q.; Shan, G.; Meng, F.; Du, D.; Wang, S.; et al. Transplantation of ACE2- Mesenchymal Stem Cells Improves the Outcome of Patients with COVID-19 Pneumonia. Aging Dis. 2020, 11, 216. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Geng, S.; Tang, S.; Yang, H.; Xiong, W.; Xu, F.; Yuan, Q.; Xiao, X.; Huang, R.; Liang, H.; et al. Clinical Efficacy and Mechanism of Mesenchymal Stromal Cells in Treatment of COVID-19. Stem Cell Res. Ther. 2022, 13, 61. [Google Scholar] [CrossRef]
- Zhu, R.; Yan, T.; Feng, Y.; Liu, Y.; Cao, H.; Peng, G.; Yang, Y.; Xu, Z.; Liu, J.; Hou, W.; et al. Mesenchymal Stem Cell Treatment Improves Outcome of COVID-19 Patients via Multiple Immunomodulatory Mechanisms. Cell Res. 2021, 31, 1244–1262. [Google Scholar] [CrossRef]
- Yousefi Dehbidi, M.; Goodarzi, N.; Azhdari, M.H.; Doroudian, M. Mesenchymal Stem Cells and Their Derived Exosomes to Combat COVID-19. Rev. Med. Virol. 2022, 32, e2281. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, V.; Sengupta, S.; Lazo, A.; Woods, P.; Nolan, A.; Bremer, N. Exosomes Derived from Bone Marrow Mesenchymal Stem Cells as Treatment for Severe COVID-19. Stem Cells Dev. 2020, 29, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Shahani, P.; Datta, I. Mesenchymal Stromal Cell Therapy for Coronavirus Disease 2019: Which? When? And How Much? Cytotherapy 2021, 23, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Kabat, M.; Bobkov, I.; Kumar, S.; Grumet, M. Trends in Mesenchymal Stem Cell Clinical Trials 2004-2018: Is Efficacy Optimal in a Narrow Dose Range? Stem Cells Transl. Med. 2020, 9, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Bahsoun, S.; Coopman, K.; Akam, E.C. The Impact of Cryopreservation on Bone Marrow-Derived Mesenchymal Stem Cells: A Systematic Review. J. Transl. Med. 2019, 17, 397. [Google Scholar] [CrossRef] [Green Version]
- Cottle, C.; Porter, A.P.; Lipat, A.; Turner-Lyles, C.; Nguyen, J.; Moll, G.; Chinnadurai, R. Impact of Cryopreservation and Freeze-Thawing on Therapeutic Properties of Mesenchymal Stromal/Stem Cells and Other Common Cellular Therapeutics. Curr. Stem Cell Rep. 2022, 8, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Laroye, C.; Gauthier, M.; Antonot, H.; Decot, V.; Reppel, L.; Bensoussan, D. Mesenchymal Stem/Stromal Cell Production Compliant with Good Manufacturing Practice: Comparison between Bone Marrow, the Gold Standard Adult Source, and Wharton’s Jelly, an Extraembryonic Source. JCM 2019, 8, 2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Vila, I.; Coca, M.I.; Grau-Vorster, M.; Pujals-Fonts, N.; Caminal, M.; Casamayor-Genescà, A.; Ortega, I.; Reales, L.; Pla, A.; Blanco, M.; et al. Evaluation of a Cell-Banking Strategy for the Production of Clinical Grade Mesenchymal Stromal Cells from Wharton’s Jelly. Cytotherapy 2016, 18, 25–35. [Google Scholar] [CrossRef]
- Yao, W.; Dong, H.; Qi, J.; Zhang, Y.; Shi, L. Safety and Efficacy of Mesenchymal Stem Cells in Severe/Critical Patients with COVID-19: A Systematic Review and Meta-Analysis. eClinicalMedicine 2022, 51, 101545. [Google Scholar] [CrossRef]
- Sion, C.; Ghannoum, D.; Ebel, B.; Gallo, F.; Isla, N.; Guedon, E.; Chevalot, I.; Olmos, E. A New Perfusion Mode of Culture for WJ-MSCs Expansion in a Stirred and Online Monitored Bioreactor. Biotech. Bioeng. 2021, 118, 4453–4464. [Google Scholar] [CrossRef]
- Avercenc-Léger, L.; Guerci, P.; Virion, J.-M.; Cauchois, G.; Hupont, S.; Rahouadj, R.; Magdalou, J.; Stoltz, J.-F.; Bensoussan, D.; Huselstein, C.; et al. Umbilical Cord-Derived Mesenchymal Stromal Cells: Predictive Obstetric Factors for Cell Proliferation and Chondrogenic Differentiation. Stem Cell Res. Ther. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Laroye, C.; Boufenzer, A.; Jolly, L.; Cunat, L.; Alauzet, C.; Merlin, J.-L.; Yguel, C.; Bensoussan, D.; Reppel, L.; Gibot, S. Bone Marrow vs Wharton’s Jelly Mesenchymal Stem Cells in Experimental Sepsis: A Comparative Study. Stem Cell Res. Ther. 2019, 10, 192. [Google Scholar] [CrossRef] [Green Version]
- Reppel, L.; Margossian, T.; Yaghi, L.; Moreau, P.; Mercier, N.; Leger, L.; Hupont, S.; Stoltz, J.-F.; Bensoussan, D.; Huselstein, C. Hypoxic Culture Conditions for Mesenchymal Stromal/Stem Cells from Wharton’s Jelly: A Critical Parameter to Consider in a Therapeutic Context. Curr. Stem Cell Res. Ther. 2014, 9, 306–318. [Google Scholar] [CrossRef]
- Abo-Aziza, F.A.M.; Zaki, A.A. The Impact of Confluence on Bone Marrow Mesenchymal Stem (BMMSC) Proliferation and Osteogenic Differentiation. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 121–132. [Google Scholar]
- Zhao, A.G.; Shah, K.; Freitag, J.; Cromer, B.; Sumer, H. Differentiation Potential of Early- and Late-Passage Adipose-Derived Mesenchymal Stem Cells Cultured under Hypoxia and Normoxia. Stem Cells Int. 2020, 2020, 8898221. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Mohamed-Ahmed, S.; Lunde, T.H.F.; Suliman, S.; Bolstad, A.I.; Hervig, T.; Mustafa, K. Influence of Platelet Storage Time on Human Platelet Lysates and Platelet Lysate-Expanded Mesenchymal Stromal Cells for Bone Tissue Engineering. Stem Cell Res. Ther. 2020, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- de Noronha, N.C.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming Approaches to Improve the Efficacy of Mesenchymal Stromal Cell-Based Therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A New Mesenchymal Stem Cell (MSC) Paradigm: Polarization into a Pro-Inflammatory MSC1 or an Immunosuppressive MSC2 Phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.-H.; Chang, C.-L.; Tsai, T.-H.; Chang, L.-T.; Leu, S.; Chen, Y.-L.; Yang, C.-C.; Chua, S.; Yeh, K.-H.; Chai, H.-T.; et al. Apoptotic Adipose-Derived Mesenchymal Stem Cell Therapy Protects against Lung and Kidney Injury in Sepsis Syndrome Caused by Cecal Ligation Puncture in Rats. Stem Cell Res. Ther. 2013, 4, 155. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.H.M.; D’Rozario, J.; Mendonca, S.; Bhuvan, T.; Payne, N.L.; Zheng, D.; Hisana, A.; Wallis, G.; Barugahare, A.; Powell, D.; et al. Mesenchymal Stromal Cell Apoptosis Is Required for Their Therapeutic Function. Nat. Commun. 2021, 12, 6495. [Google Scholar] [CrossRef]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Calfee, C.S.; Zhuo, H.; Thompson, B.T.; Wilson, J.G.; Levitt, J.E.; Rogers, A.J.; Gotts, J.E.; Wiener-Kronish, J.P.; Bajwa, E.K.; et al. Treatment with Allogeneic Mesenchymal Stromal Cells for Moderate to Severe Acute Respiratory Distress Syndrome (START Study): A Randomised Phase 2a Safety Trial. Lancet Respir. Med. 2019, 7, 154–162. [Google Scholar] [CrossRef]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Continuous | Reception | P0 | P1 | P2 | P3 | |
| Donor infectious markers | Bacteriology CFU-F Cell phenotype Count/Viability | Bacteriology Cell phenotype Karyotype CFU-F Count/Viability Telomerase activity MLR Osteocyte and adipocyte differentiation (first 3 batches) | ||||
| Discontinuous | Reception | P0 | P1 | P2 | Cryopreservation/Thawing | P3 |
| Donor infectious markers | Bacteriology CFU-F Cell phenotype Count/Viability | Bacteriology Count/Viability | Bacteriology Cell phenotype Karyotype CFU-F Count/Viability Telomerase activity MLR Osteocyte and adipocyte differentiation (first 3 batches) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laroye, C.; Gauthier, M.; Morello, J.; Charif, N.; Cannard, V.L.; Bonnet, C.; Lozniewski, A.; Tchirkov, A.; De Isla, N.; Decot, V.; et al. Scale-Up of Academic Mesenchymal Stromal Cell Production. J. Clin. Med. 2023, 12, 4414. https://doi.org/10.3390/jcm12134414
Laroye C, Gauthier M, Morello J, Charif N, Cannard VL, Bonnet C, Lozniewski A, Tchirkov A, De Isla N, Decot V, et al. Scale-Up of Academic Mesenchymal Stromal Cell Production. Journal of Clinical Medicine. 2023; 12(13):4414. https://doi.org/10.3390/jcm12134414
Chicago/Turabian StyleLaroye, Caroline, Mélanie Gauthier, Jessica Morello, Naceur Charif, Véronique Latger Cannard, Céline Bonnet, Alain Lozniewski, Andrei Tchirkov, Natalia De Isla, Véronique Decot, and et al. 2023. "Scale-Up of Academic Mesenchymal Stromal Cell Production" Journal of Clinical Medicine 12, no. 13: 4414. https://doi.org/10.3390/jcm12134414