

Potential Role of VHL, PTEN, and BAP1 Mutations in Renal Tumors
 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Preparation of Renal Tumor Tissue Samples from Patients
2.2. RNA Isolation and Reverse Transcription PCR
2.3. Tissue Sample Preparation and DNA Isolation
2.4. PCR Amplification Prior to Sequencing
- A.
- for VHL
- B.
- for PTEN
- C.
- for BAP1
2.5. DNA Sequencing
3. Results
3.1. Clinicopathological Characteristics of the Patients
3.2. Expression of mRNA for BAP1 and PTEN in Human Kidney Tumor Tissue Samples
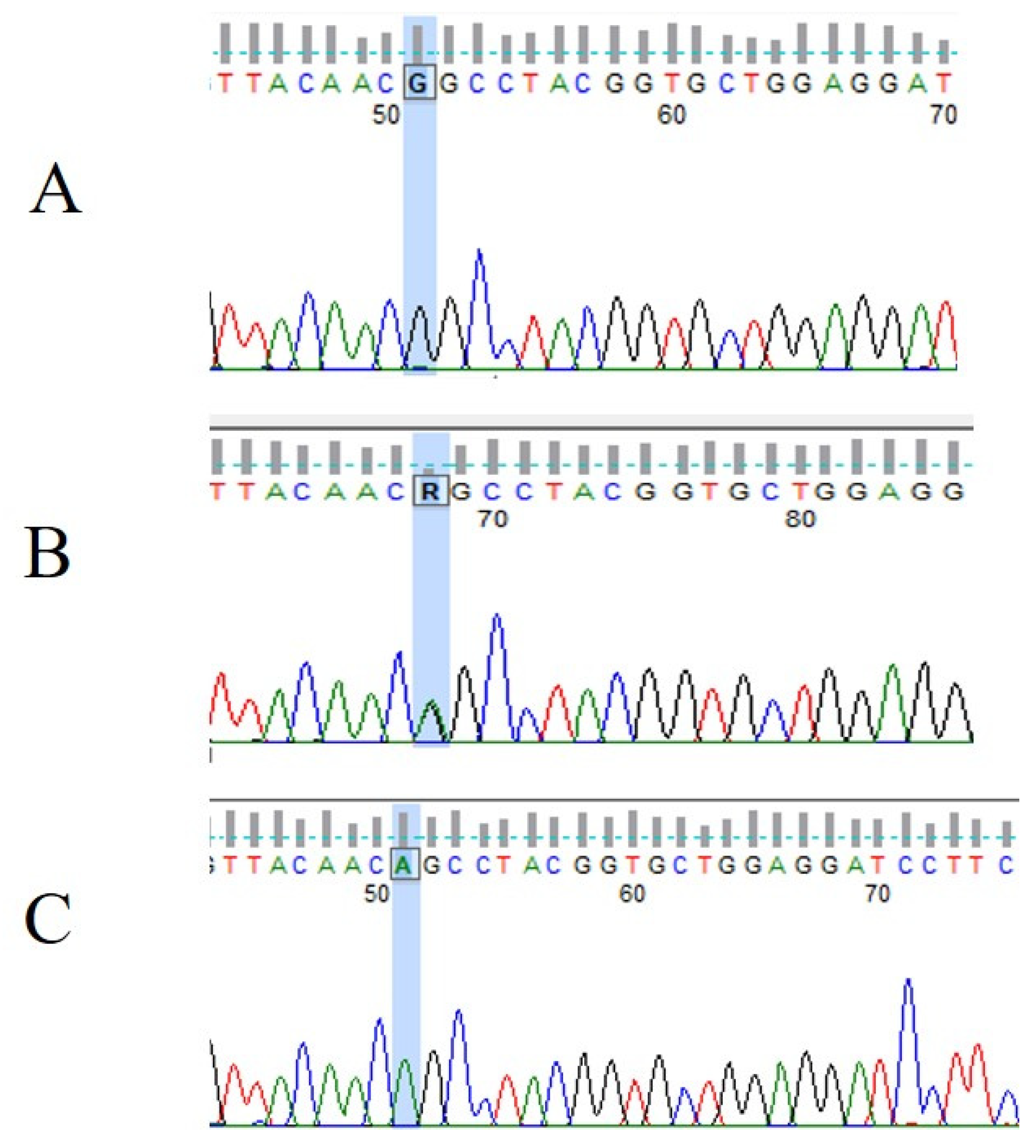
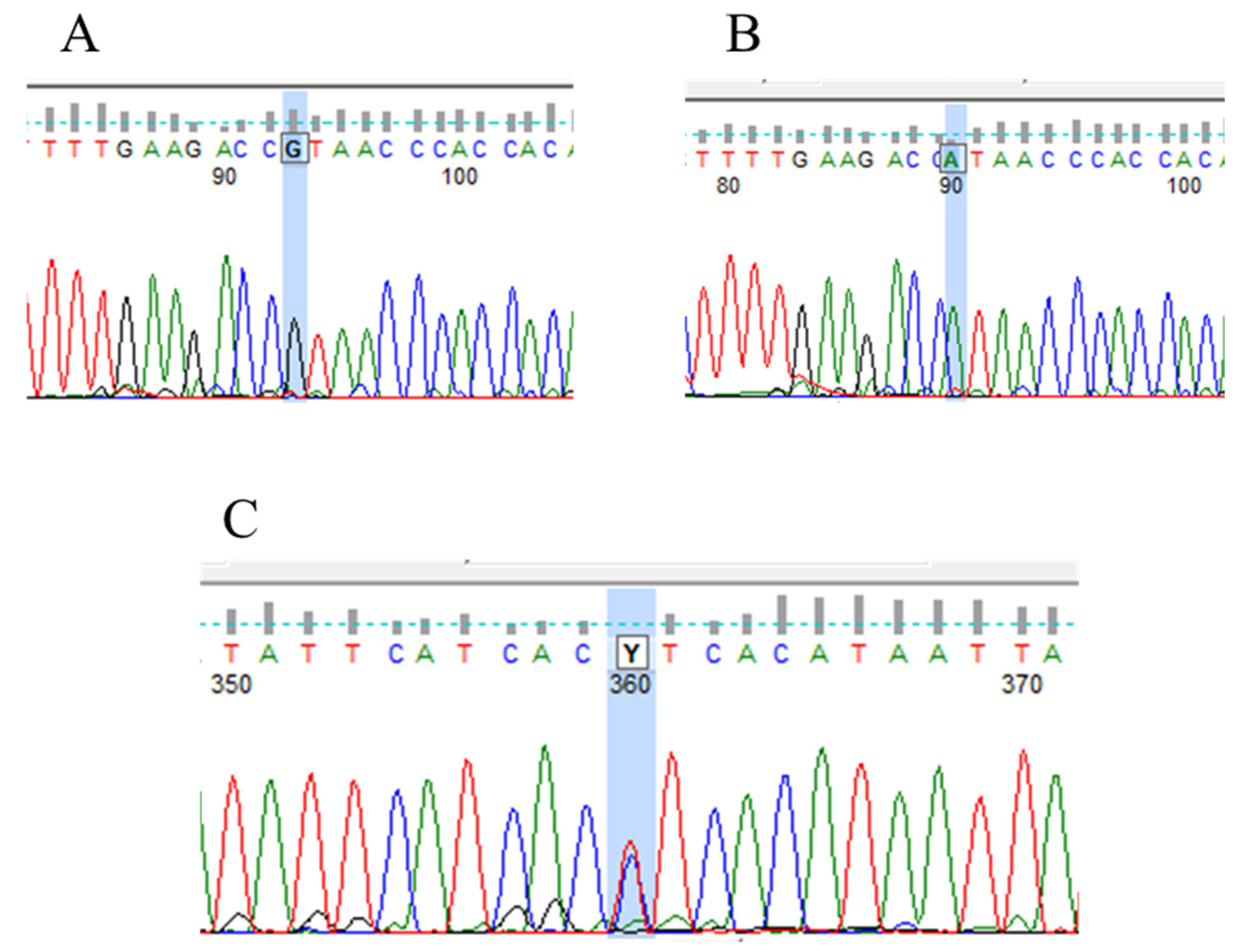
3.3. VHL Mutations and IVS1-195 Nt G/A Polymorphism in Human Renal Tumor Specimens Examined
3.4. PTEN Mutations and Polymorphism in Human Renal Tumor Specimens Examined
3.5. Wild Types of the BAP1 Gene in Human Renal Tumor Specimens Examined
3.6. Relationship between VHL, PTEN, and BAP1 Mutations and Clinicopathological Features of the Human Specimens Examined
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bukowski, R.M. Prognostic factors for survival in metastatic renal cell carcinoma: Update 2008. Cancer 2009, 115, 2273–2281. [Google Scholar] [CrossRef]
- Kumar, A.; Kumari, N.; Gupta, V.; Prasad, R. Renal Cell Carcinoma: Molecular Aspects. Indian J. Clin. Biochem. IJCB 2018, 33, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xi, Z.; Xi, J.; Zhang, H.; Li, J.; Xia, Y.; Yi, Y. Somatic mutations in renal cell carcinomas from Chinese patients revealed by whole exome sequencing. Cancer Cell Int. 2018, 18, 159. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.R.; Surabhi, V.R.; Menias, C.O.; Raut, A.A.; Chintapalli, K.N. Benign renal neoplasms in adults: Cross-sectional imaging findings. AJR Am. J. Roentgenol. 2008, 190, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Younus, M.S.; Stephen, W.L. Renal Angiomyolipoma. StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK585104/ (accessed on 24 May 2023).
- Muscarella, L.A.; D’Agruma, L.; la Torre, A.; Gigante, M.; Coco, M.; Parrella, P.; Battaglia, M.; Carrieri, G.; Carella, M.; Zelante, L.; et al. VHL gene alterations in Italian patients with isolated renal cell carcinomas. Int. J. Biol. Markers 2013, 28, 208–215. [Google Scholar] [CrossRef]
- Dandanell, M.; Friis-Hansen, L.; Sunde, L.; Nielsen, F.C.; Hansen, T.V. Identification of 3 novel VHL germ-line mutations in Danish VHL patients. BMC Med. Genet. 2012, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G., Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL−/− clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, J.H.; Jang, H.J.; Han, B.; Zang, D.Y. Clinicopathologic Significance of VHL Gene Alteration in Clear-Cell Renal Cell Carcinoma: An Updated Meta-Analysis and Review. Int. J. Mol. Sci. 2018, 19, 2529. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.Z.; Xu, L.W.; Zhou, C.C.; Lu, T.Z.; Yao, W.T.; Wu, R.; Zhao, Y.C.; Xu, X.; Hu, Z.K.; Wang, M.; et al. A BAP1 Mutation-specific MicroRNA Signature Predicts Clinical Outcomes in Clear Cell Renal Cell Carcinoma Patients with Wild-type BAP1. J. Cancer 2017, 8, 2643–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, S.; Gardie, B.; Couvé, S.; Gad, S. Von Hippel-Lindau: How a rare disease illuminates cancer biology. Semin. Cancer Biol. 2013, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Crespigio, J.; Berbel, L.C.L.; Dias, M.A.; Berbel, R.F.; Pereira, S.S.; Pignatelli, D.; Mazzuco, T.L. Von Hippel-Lindau disease: A single gene, several hereditary tumors. J. Endocrinol. Investig. 2018, 41, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Venkatesh, K.; Srikanth, L.; Sarma, P.V.; Reddy, A.R.; Subramanian, S.; Phaneendra, B.V. Novel three missense mutations observed in Von Hippel-Lindau gene in a patient reported with renal cell carcinoma. Indian J. Hum. Genet. 2013, 19, 373–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minardi, D.; Lucarini, G.; Milanese, G.; Montironi, R.; Di Primio, R. Prognostic role of BAP1 in pT1 clear cell carcinoma in partial nephrectomy specimens. Virchows Arch. 2017, 471, 99–105. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Nasu, M.; Emi, M.; Pastorino, S.; Tanji, M.; Powers, A.; Luk, H.; Baumann, F.; Zhang, Y.A.; Gazdar, A.; Kanodia, S.; et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J. Thorac. Oncol. 2015, 10, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-d’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Zhao, C.; Li, S.; Wang, J.; Zhou, Q.; Sun, J.; Ding, Q.; Liu, M.; Ding, G. EZH2 Expression is increased in BAP1-mutant renal clear cell carcinoma and is related to poor prognosis. J. Cancer 2018, 9, 3787–3796. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Zhou, S.F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Dev. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Giudice, F.S.; Squarize, C.H. The determinants of head and neck cancer: Unmasking the PI3K pathway mutations. J. Carcinog. Mutagen. 2013, S5, 3. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Longy, M. Inherited PTEN mutations and the prediction of phenotype. Semin. Cell Dev. Biol. 2016, 52, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Kim, M.J.; Jeon, H.S.; Choi, J.E.; Kim, D.S.; Lee, E.B.; Cha, S.I.; Yoon, G.S.; Kim, C.H.; Jung, T.H.; et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer 2010, 69, 279–283. [Google Scholar] [CrossRef]
- Lynch, E.D.; Ostermeyer, E.A.; Lee, M.K.; Arena, J.F.; Ji, H.; Dann, J.; Swisshelm, K.; Suchard, D.; MacLeod, P.M.; Kvinnsland, S.; et al. Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am. J. Hum. Genet. 1997, 61, 1254–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh Durban, V.; Jansen, M.; Davies, E.J.; Morsink, F.H.; Offerhaus, G.J.; Clarke, A.R. Epithelial-specific loss of PTEN results in colorectal juvenile polyp formation and invasive cancer. Am. J. Pathol. 2014, 184, 86–91. [Google Scholar] [CrossRef]
- Minaguchi, T.; Yoshikawa, H.; Oda, K.; Ishino, T.; Yasugi, T.; Onda, T.; Nakagawa, S.; Matsumoto, K.; Kawana, K.; Taketani, Y. PTEN mutation located only outside exons 5, 6, and 7 is an independent predictor of favorable survival in endometrial carcinomas. Clin. Cancer Res. 2001, 7, 2636–2642. [Google Scholar]
- Yang, J.; Ren, Y.; Wang, L.; Li, B.; Chen, Y.; Zhao, W.; Xu, W.; Li, T.; Dai, F. PTEN mutation spectrum in breast cancers and breast hyperplasia. J. Cancer Res. Clin. Oncol. 2010, 136, 1303–1311. [Google Scholar] [CrossRef]
- Aguissa-Touré, A.H.; Li, G. Genetic alterations of PTEN in human melanoma. Cell Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef]
- Baig, R.M.; Mahjabeen, I.; Sabir, M.; Masood, N.; Hafeez, S.; Malik, F.A.; Kayani, M.A. Genetic changes in the PTEN gene and their association with breast cancer in Pakistan. Asian Pac. J. Cancer Prev. 2011, 12, 2773–2778. [Google Scholar]
- Malentacchi, F.; Turrini, I.; Sorbi, F.; Projetto, E.; Castiglione, F.; Fambrini, M.; Petraglia, F.; Pillozzi, S.; Noci, I. Pilot investigation of the mutation profile of PIK3CA/PTEN genes (PI3K pathway) in grade 3 endometrial cancer. Oncol. Rep. 2019, 41, 1560–1574. [Google Scholar] [CrossRef]
- Wen, Y.G.; Wang, Q.; Zhou, C.Z.; Qiu, G.Q.; Peng, Z.H.; Tang, H.M. Mutation analysis of tumor suppressor gene PTEN in patients with gastric carcinomas and its impact on PI3K/AKT pathway. Oncol. Rep. 2010, 24, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Liu, C.X.; Nong, W.X.; Chen, Y.Z.; Qi, Y.; Li, H.A.; Hu, W.H.; Sun, K.; Li, F. Mutational analysis of p53 and PTEN in soft tissue sarcoma. Mol. Med. Rep. 2012, 5, 457–461. [Google Scholar] [CrossRef]
- Mattocks, C.; Tarpey, P.; Bobrow, M.; Whittaker, J. Comparative sequence analysis (CSA): A new sequence-based method for the identification and characterization of mutations in DNA. Hum. Mutat. 2000, 16, 437–443. [Google Scholar] [CrossRef]
- Yoshida, M.; Ashida, S.; Kondo, K.; Kobayashi, K.; Kanno, H.; Shinohara, N.; Shitara, N.; Kishida, T.; Kawakami, S.; Baba, M.; et al. Germ-line mutation analysis in patients with von Hippel-Lindau disease in Japan: An extended study of 77 families. Jpn. J. Cancer Res. 2000, 91, 204–212. [Google Scholar] [CrossRef]
- Chen, F.; Kishida, T.; Yao, M.; Hustad, T.; Glavac, D.; Dean, M.; Gnarra, J.R.; Orcutt, M.L.; Duh, F.M.; Glenn, G.; et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum. Mutat. 1995, 5, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, E.; Martella, M.; Tosatto, S.C.; Murgia, A. Identification and in silico analysis of novel von Hippel-Lindau (VHL) gene variants from a large population. Ann. Hum. Genet. 2011, 75, 483–496. [Google Scholar] [CrossRef]
- Whaley, J.M.; Naglich, J.; Gelbert, L.; Hsia, Y.E.; Lamiell, J.M.; Green, J.S.; Collins, D.; Neumann, H.P.; Laidlaw, J.; Li, F.P.; et al. Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene are similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma. Am. J. Hum. Genet. 1994, 55, 1092–1102. [Google Scholar] [PubMed]
- Lv, C.; Bai, Z.; Liu, Z.; Luo, P.; Zhang, J. Renal cell carcinoma risk is associated with the interactions of APOE, VHL and MTHFR gene polymorphisms. Int. J. Clin. Exp. Pathol. 2015, 8, 5781–5786. [Google Scholar]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wang, X.; Tang, Z.; Qiu, X.; Guo, Z.; Huang, D.; Xiong, H.; Guo, Q. The Correlation Between Tuberous Sclerosis Complex Genotype and Renal Angiomyolipoma Phenotype. Front. Genet. 2020, 11, 575750. [Google Scholar] [CrossRef]
- Song, X.; Peng, Y.; Wang, X.; Chen, Y.; Jin, L.; Yang, T.; Qian, M.; Ni, W.; Tong, X.; Lan, J. Incidence, Survival, and Risk Factors for Adults with Acute Myeloid Leukemia Not Otherwise Specified and Acute Myeloid Leukemia with Recurrent Genetic Abnormalities: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database, 2001–2013. Acta Haematol. 2018, 139, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Fiske, J.; Patel, R.; Kau, E.; Pappas, J.G.; Garcia, R.A.; Taneja, S.S. Multifocal renal oncocytoma in a patient with Von Hippel-Lindau mutation. Urology 2005, 66, 1320. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Serie, D.J.; Cheville, J.C.; Ho, T.H.; Kapur, P.; Brugarolas, J.; Thompson, R.H.; Leibovich, B.C.; Kwon, E.D.; Joseph, R.W.; et al. BAP1 and PBRM1 in metastatic clear cell renal cell carcinoma: Tumor heterogeneity and concordance with paired primary tumor. BMC Urol. 2017, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Farley, M.N.; Schmidt, L.S.; Mester, J.L.; Pena-Llopis, S.; Pavia-Jimenez, A.; Christie, A.; Vocke, C.D.; Ricketts, C.J.; Peterson, J.; Middelton, L.; et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol. Cancer Res. 2013, 11, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Brenner, W.; Färber, G.; Herget, T.; Lehr, H.A.; Hengstler, J.G.; Thüroff, J.W. Loss of tumor suppressor protein PTEN during renal carcinogenesis. Int. J. Cancer 2002, 99, 53–57. [Google Scholar] [CrossRef]
- Wang, S.S.; Gu, Y.F.; Wolff, N.; Stefanius, K.; Christie, A.; Dey, A.; Hammer, R.E.; Xie, X.J.; Rakheja, D.; Pedrosa, I.; et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 16538–16543. [Google Scholar] [CrossRef]
- Teh, B.T.; Blennow, E.; Giraud, S.; Sahlén, S.; Hii, S.I.; Brookwell, R.; Brauch, H.; Nordenskjöld, M.; Larsson, C.; Nicol, D. Bilateral multiple renal oncocytomas and cysts associated with a constitutional translocation (8;9)(q24.1;q34.3) and a rare constitutional VHL missense substitution. Genes Chromosomes Cancer 1998, 21, 260–264. [Google Scholar] [CrossRef]
- Chrabańska, M.; Szweda-Gandor, N.; Drozdzowska, B. Two Single Nucleotide Polymorphisms in the Von Hippel-Lindau Tumor Suppressor Gene in Patients with Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 3778. [Google Scholar] [CrossRef]
- Webster, B.R.; Gopal, N.; Ball, M.W. Tumorigenesis Mechanisms Found in Hereditary Renal Cell Carcinoma: A Review. Genes 2022, 13, 2122. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Pandith, A.A.; Shah, Z.A.; Wani, M.S. The Profound Impact of von Hippel-Lindau Gene Mutations in Renal Cell Cancers: A Study of the Kashmiri Population. UroToday Int. J. 2013, 6, 14. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Age/Gender | Histological Type | Histological Grade | Pathological Stage | TNM Classification |
|---|---|---|---|---|---|
| 1. | 40/female | ccRCC | 2 | pT1a | T1, N0, M0 |
| 2. | 68/female | ccRCC | 2 | pT4 | T4, N0, M0 |
| 3. | 62/male | ccRCC | 3 | pT4 | T4, N0, M0 |
| 4. | 64/male | ccRCC | 3 | pT4 | T4, N0, M0 |
| 5. | 72/female | ccRCC | 3 | pT3a | T3, N0, M0 |
| 6. | 55/male | ccRCC | 2 | pT1a | T1, N0, M0 |
| 7. | 57/female | ccRCC | 2 | pT1a | T1, N0, M0 |
| 8. | 36/male | ccRCC | 3 | pT4 | T4, N0, M0 |
| 9. | 39/female | ccRCC | 3 | pT1a | T1, N0, M0 |
| 10. | 72/female | AML | ― | ― | ― |
| 11. | 72/female | oncocytoma | ― | ― | ― |
| 12. | 47/female | ccRCC | 3 | pT1a | T1, N0, M0 |
| 13. | 69/male | ccRCC | 3 | pT4 | T4, N0, M0 |
| 14. | 53/female | ccRCC | 3 | pT4 | T4, N0, M0 |
| 15. | 49/male | ccRCC | 3 | pT1a | T1, N0, M0 |
| 16. | 52/male | ccRCC | 3 | pT1b | T1, N0, M0 |
| 17. | 56/male | ccRCC | 3 | pT1b | T1,Nx, Mx |
| 18. | 69/male | ccRCC | 3 | pT1a | T1, N0, M0 |
| 19. | 37/female | ccRCC | 3 | pT1b | T1, N0, M0 |
| 20. | 49/male | ccRCC | 2 | pT1a | T1, N0, M0 |
| 21. | 72/female | ccRCC | 2 | pT1a | T1, N0, M0 |
| 22. | 36/female | AML | ― | ― | ― |
| 23. | 41/male | ccRCC | 2 | pT1b | T1, N0, M0 |
| 24. | 46/male | pRCC | 2 | pT1b | T1, N0, M0 |
| Histological Grade | Grade % | Men Number % | Women Number % |
|---|---|---|---|
| Grade 2 | 8 cases (38.095%) | 4 50.0 | 4 50.0 |
| Grade 3 | 13 cases (61.90%) | 8 61.53 | 5 48.46 |
| Sample Number | Age/Gender | Histological Type | Histological Grade | Pathological Stage | TNM Clinical Stage | Tumor Size | Surgery Type | Mutation | Other Tumors |
|---|---|---|---|---|---|---|---|---|---|
| 1. | 72/female | Oncocytoma | ― | ― | ― | 10 cm | right site radical nephrectomy | VHL mutation | was not observed |
| 2. | 57/female | ccRCC | 2 | pT1a | T1, N0, M0 | 3.3–4 cm | right site kidney nephrectomy | VHL mutation and VHL polymorphism | was not observed |
| 3. | 36/male | ccRCC | 3 | pT4 | T4, N0, M0 | 4 cm | right site kidney resection | PTEN polymorphism | was not observed |
| 4. | 39/female | AML | ― | ― | ― | 3.3 cm | right site kidney laparoscopic nephrectomy | VHL mutation | was not observed |
| 5. | 37/female | ccRCC | 3 | pT1b | T1, N0, M0 | 6 cm | left site kidney resection | VHL mutation | was not observed |
| * 6. | 72/female | ccRCC | 2 | pT1a | T1, N0, M0 | 2 × 1.5 cm | right site laparoscopic kidney nephrectomy | VHL mutation | cervical carcinoma |
| 7. | 36/female | AML | ― | ― | ― | 3.3 cm | right site kidney resection | VHL mutation | bladder cancer |
| 8. | 46/male | pRCC | 2 | pT1b | T1, N0, M0 | 5 cm | right site kidney resection, open surgery | PTEN mutation | was not observed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szegedi, K.; Szabó, Z.; Kállai, J.; Király, J.; Szabó, E.; Bereczky, Z.; Juhász, É.; Dezső, B.; Szász, C.; Zsebik, B.; et al. Potential Role of VHL, PTEN, and BAP1 Mutations in Renal Tumors. J. Clin. Med. 2023, 12, 4538. https://doi.org/10.3390/jcm12134538
Szegedi K, Szabó Z, Kállai J, Király J, Szabó E, Bereczky Z, Juhász É, Dezső B, Szász C, Zsebik B, et al. Potential Role of VHL, PTEN, and BAP1 Mutations in Renal Tumors. Journal of Clinical Medicine. 2023; 12(13):4538. https://doi.org/10.3390/jcm12134538
Chicago/Turabian StyleSzegedi, Krisztián, Zsuzsanna Szabó, Judit Kállai, József Király, Erzsébet Szabó, Zsuzsanna Bereczky, Éva Juhász, Balázs Dezső, Csaba Szász, Barbara Zsebik, and et al. 2023. "Potential Role of VHL, PTEN, and BAP1 Mutations in Renal Tumors" Journal of Clinical Medicine 12, no. 13: 4538. https://doi.org/10.3390/jcm12134538
APA StyleSzegedi, K., Szabó, Z., Kállai, J., Király, J., Szabó, E., Bereczky, Z., Juhász, É., Dezső, B., Szász, C., Zsebik, B., Flaskó, T., & Halmos, G. (2023). Potential Role of VHL, PTEN, and BAP1 Mutations in Renal Tumors. Journal of Clinical Medicine, 12(13), 4538. https://doi.org/10.3390/jcm12134538