
Epithelioid Sarcoma of the Spine: A Review of Literature and Case Report
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Epithelioid Sarcoma: Natural History and Presentation
1.2. Epithelioid Sarcoma: Histopathology
1.3. Epithelioid Sarcoma: Diagnosis
1.4. Epithelioid Sarcoma: Evaluation, Treatment, and Prognosis
1.5. Epithelioid Sarcoma of the Spine
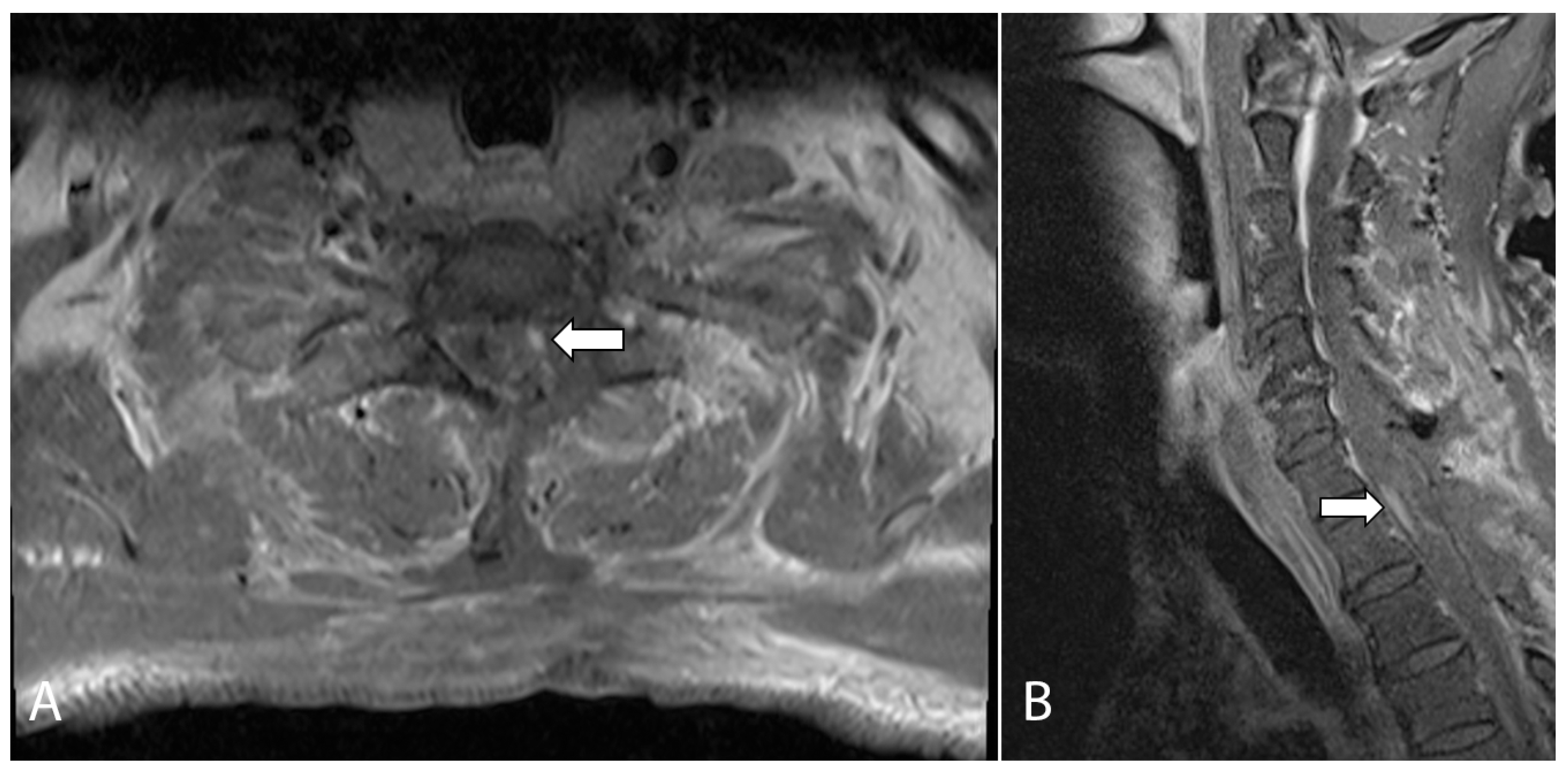
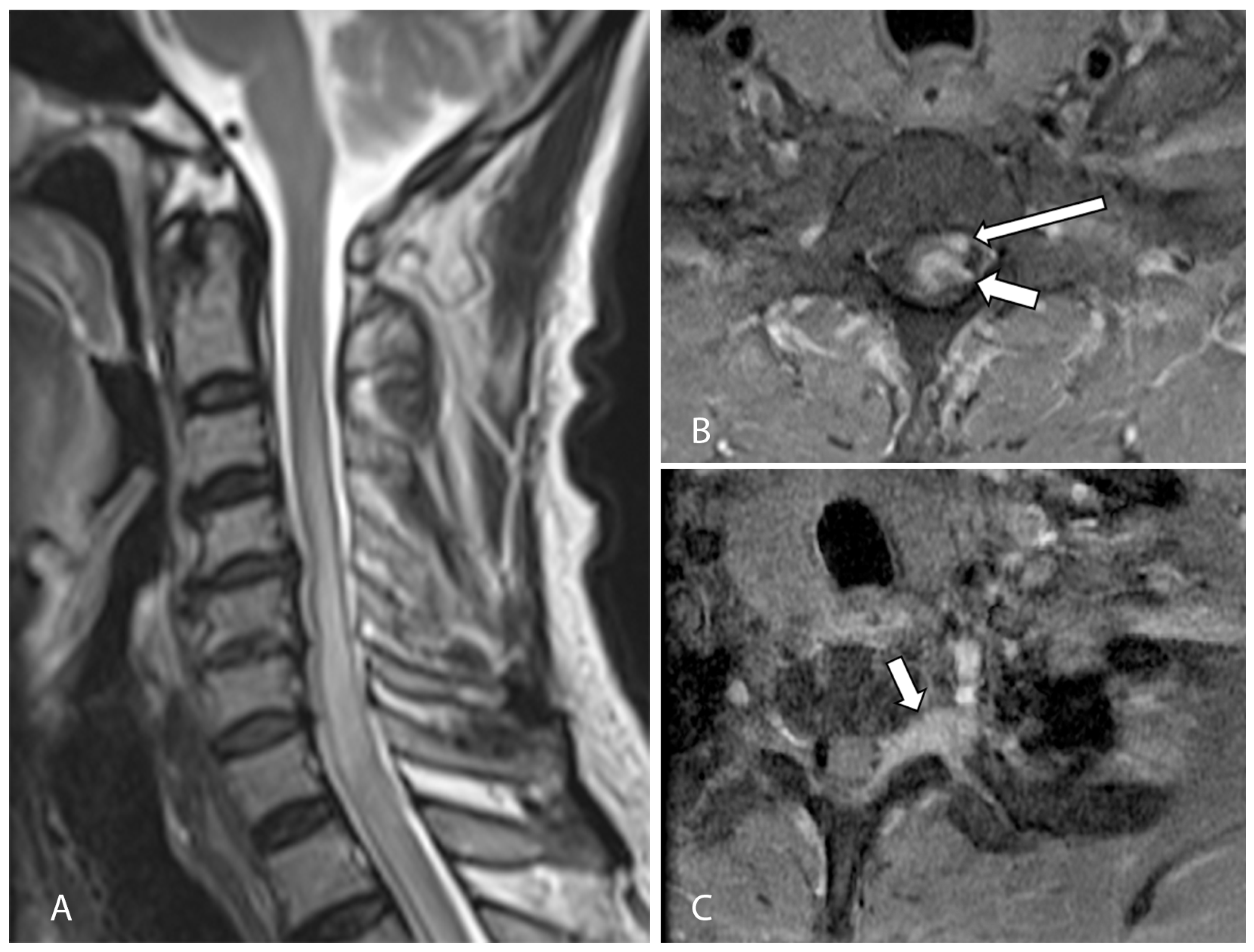
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jawad, M.U.; Extein, J.; Min, E.S.; Scully, S.P. Prognostic Factors for Survival in Patients with Epithelioid Sarcoma: 441 Cases from the SEER Database. Clin. Orthop. Relat. Res. 2009, 467, 2939–2948. [Google Scholar] [CrossRef]
- Enzinger, F.M. Epithelioid Sarcoma.A Sarcoma Simulating a Granuloma or a Carcinoma. Cancer 1970, 26, 1029–1041. [Google Scholar] [CrossRef]
- Akhtar, K.; Piyush, A.; Haiyat, S.; Khan, A. Epithelioid Sarcoma: A Diagnostic Challenge of a Rare Presentation. Arch. Int. Surg. 2016, 6, 57. [Google Scholar] [CrossRef]
- Dion, E.; Forest, M.; Brasseur, J.-L.; Amoura, Z.; Grenier, P. Epithelioid Sarcoma Mimicking Abscess: Review of the MRI Appearances. Skelet. Radiol. 2001, 30, 173–177. [Google Scholar] [CrossRef]
- Fisher, C. Epithelioid Sarcoma of Enzinger. Adv. Anat. Pathol. 2006, 13, 114–121. [Google Scholar] [CrossRef]
- Weisskopf, M.; Münker, R.; Hermanns-Sachweh, B.; Ohnsorge, J.A.K.; Siebert, C. Epithelioid Sarcoma in the Thoracic Spine. Eur. Spine J. 2006, 15, 604–609. [Google Scholar] [CrossRef]
- Yoo, J.H. Epithelioid Sarcoma of the Spine: A Case Report and Review. Open J. Clin. Diagn. 2011, 1, 1–4. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Bellan, E.; Dei Tos, A.P. The 2020 WHO Classification of Soft Tissue Tumours: News and Perspectives. Pathologica 2020, 113, 70–84. [Google Scholar] [CrossRef]
- Del Savio, E.; Maestro, R. Beyond SMARCB1 Loss: Recent Insights into the Pathobiology of Epithelioid Sarcoma. Cells 2022, 11, 2626. [Google Scholar] [CrossRef]
- Chase, D.R.; Enzinger, F.M. Epithelioid sarcoma: Diagnosis, prognostic indicators, and treatment. Am. J. Surg. Pathol. 1985, 9, 241–263. [Google Scholar] [CrossRef]
- Armah, H.B.; Parwani, A.V. Epithelioid Sarcoma. Arch. Pathol. Lab. Med. 2009, 133, 814–819. [Google Scholar] [CrossRef]
- Pendse, A.A.; Dodd, L.G. Fine-Needle-Aspiration Cytology of a Proximal Type Epithelioid Sarcoma: A Case Report. Diagn. Cytopathol. 2015, 43, 859–862. [Google Scholar] [CrossRef]
- Kim, C.; Yoo, K.H.; Kim, M.H.; Chon, H.J.; Lee, S.I.; Lee, H.J.; Koh, S.; Lee, H.Y.; Lee, H.R.; Kim, K.S.; et al. Different Subtypes of Epithelioid Sarcoma and Their Clinical Implication: Long-Term Multi-Institutional Experience with a Rare Sarcoma. APMIS 2017, 125, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L. Selected Topics in the Pathology of Epithelioid Soft Tissue Tumors. Mod. Pathol. 2014, 27, S64–S79. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Sobczuk, P.; Kostrzanowski, M.; Spalek, M.; Chojnacka, M.; Szumera-Cieckiewicz, A.; Rutkowski, P. Epithelioid Sarcoma—From Genetics to Clinical Practice. Cancers 2020, 12, 2112. [Google Scholar] [CrossRef] [PubMed]
- Chbani, L.; Guillou, L.; Terrier, P.; Decouvelaere, A.V.; Grégoire, F.; Terrier-Lacombe, M.J.; Ranchère, D.; Robin, Y.M.; Collin, F.; Fréneaux, P.; et al. Epithelioid Sarcoma. Am. J. Clin. Pathol. 2009, 131, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Dal Cin, P.; Fletcher, C.D.M. Loss of INI1 Expression Is Characteristic of Both Conventional and Proximal-Type Epithelioid Sarcoma. Am. J. Surg. Pathol. 2009, 33, 542–550. [Google Scholar] [CrossRef]
- Miettinen, M.; Fanburg-Smith, J.C.; Virolainen, M.; Shmookler, B.M.; Fetsch, J.F. Epithelioid Sarcoma: An Immunohistochemical Analysis of 112 Classical and Variant Cases and a Discussion of the Differential Diagnosis. Hum. Pathol. 1999, 30, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Callister, M.D.; Ballo, M.T.; Pisters, P.W.T.; Patel, S.R.; Feig, B.W.; Pollock, R.E.; Benjamin, R.S.; Zagars, G.K. Epithelioid Sarcoma: Results of Conservative Surgery and Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 384–391. [Google Scholar] [CrossRef] [PubMed]
- De Visscher, S.A.H.J.; van Ginkel, R.J.; Wobbes, T.; Veth, R.P.H.; ten Heuvel, S.E.; Suurmeijer, A.J.H.; Hoekstra, H.J. Epithelioid Sarcoma: Still an Only Surgically Curable Disease. Cancer 2006, 107, 606–612. [Google Scholar] [CrossRef]
- Baratti, D.; Pennacchioli, E.; Casali, P.G.; Bertulli, R.; Lozza, L.; Olmi, P.; Collini, P.; Radaelli, S.; Fiore, M.; Gronchi, A. Epithelioid Sarcoma: Prognostic Factors and Survival in a Series of Patients Treated at a Single Institution. Ann. Surg. Oncol. 2007, 14, 3542–3551. [Google Scholar] [CrossRef]
- Li, Y.; Cao, G.; Tao, X.; Guo, J.; Wu, S.; Tao, Y. Clinicopathologic Features of Epithelioid Sarcoma: Report of Seventeen Cases and Review of Literature. Int. J. Clin. Exp. Pathol. 2019, 12, 3042–3048. [Google Scholar]
- Ross, H.M.; Lewis, J.J.; Woodruff, J.M.; Brennan, M.F. Epithelioid Sarcoma: Clinical Behavior and Prognostic Factors of Survival. Ann. Surg. Oncol. 1997, 4, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Elsamna, S.T.; Amer, K.; Elkattawy, O.; Beebe, K.S. Epithelioid Sarcoma: Half a Century Later. Acta Oncol. 2020, 59, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Shimm, D.S.; Suit, H.D. Radiation Therapy of Epithelioid Sarcoma. Cancer 1983, 52, 1022–1025. [Google Scholar] [CrossRef]
- Hu, W.; Wu, X.; Ma, H.; Wang, H.; Shi, X.; Zhang, K.; Gao, Y. Systematic Review of Published Cases of Primary Epithelioid Sarcoma of the Spine. Med. Sci. Monit. 2022, 29, e938416. [Google Scholar] [CrossRef]
- Babu, R.; Karikari, I.O.; Cummings, T.J.; Gottfried, O.N.; Bagley, C.A. Treatment and Outcomes of Epithelioid Sarcoma of the Spine. J. Clin. Neurosci. 2013, 20, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Chanplakorn, P.; Chanplakorn, N.; Pongtippan, A.; Jaovisidha, S.; Laohacharoensombat, W. Recurrent Epithelioid Sarcoma in the Thoracic Spine Successfully Treated with Multilevel Total En Bloc Spondylectomy. Eur. Spine J. 2011, 20, 302–308. [Google Scholar] [CrossRef]
- Steib, J.-P.; Pierchon, F.; Farcy, J.P.; Lang, G.; Christmann, D.; Gnassia, J.-P. Epithelioid Sarcoma of the Spine: A Case Report. Spine 1996, 21, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A. Targeting Epigenetics in Sarcomas through EZH2 Inhibition. J. Hematol. Oncol. 2020, 13, 33. [Google Scholar] [CrossRef]
- Wang, J.; Lu, C.; Tang, X. Response to Immunotherapy in a Patient with Advanced Epithelioid Sarcoma of Adrenal Gland: A Case Report. Exp. Ther. Med. 2022, 24, 659. [Google Scholar] [CrossRef] [PubMed]
- Pecora, A.; Halpern, S.; Weber, M.; Paleoudis, E.G.; Panush, D.; Patterson, F.; Toretsky, J. Rapid and Complete Response to Combination Anti-CTLA-4 and Anti-PD-1 Checkpoint Inhibitor Therapy in a Patient With Stage IV Refractory End-Stage Epithelioid Sarcoma: A Case Report. J. Immunother. 2020, 43, 286–290. [Google Scholar] [CrossRef]
- Kashyap, D.; Rastogi, S.; Garg, V.; Shrivastava, S.; Barwad, A.; Shamim, S.A.; Hemrom, A.; Dhamija, E.; Bhoriwal, S.; Garg, R. Epithelioid Sarcoma and Its Outcome: A Retrospective Analysis from a Tertiary Care Center in North India. Future Sci. OA 2022, 8, FSO822. [Google Scholar] [CrossRef]
- Sudhir, G.; Jayabalan, S.; Ram, A.; Gadde, S.; Kailash, K. Epithelioid Sarcoma of Lumbar Spine: A Rare Mesenchymal Tumor Masquerading as Infection. Asian J. Neurosurg. 2021, 16, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Chamadoira, C.; Pereira, P.; Silva, P.S.; Castro, L.; Vaz, R. Epithelioid Sarcoma of the Spine: Case Report and Literature Review. Neurocirugía 2014, 25, 179–182. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Huang, M.-Y.; Chang, C.-C.; Lee, C.-S.; Liao, Y.-M.; Chiu, S.-S.; Chang, T.-T. FDG PET/CT Findings of Epithelioid Sarcoma in a Pediatric Patient. Clin. Nucl. Med. 2007, 32, 898–901. [Google Scholar] [CrossRef]
- Bydon, M.; De la Garza-Ramos, R.; Suk, I.; McCarthy, E.; Yamada, Y.; Wolinsky, J.-P.; Gokaslan, Z.L. Single-Staged Multilevel Spondylectomy for En Bloc Resection of an Epithelioid Sarcoma with Intradural Extension in the Cervical Spine: Technical Case Report. Oper. Neurosurg. 2015, 11, E585–E593. [Google Scholar] [CrossRef]
- Kurtkaya-Yapıcıer, Ö.; Dedrick, D.J. Primary Epithelioid Sarcoma of the Dura: Case Report. Neurosurgery 2002, 50, 198–203. [Google Scholar] [CrossRef]
- Baardsen, E.; Thomas, S.; Suzuki, G. A Rare Case of Primary Intracranial Epithelioid Sarcoma in a Patient with Neurofibromatosis Type 2. Am. J. Clin. Pathol. 2016, 146, 63. [Google Scholar] [CrossRef]
- Haider, A.S.; Palmisciano, P.; Sagoo, N.S.; Bin Alamer, O.; El Ahmadieh, T.Y.; Pan, E.; Garzon-Muvdi, T. Primary Central Nervous System Sarcomas in Adults: A Systematic Review. Clin. Neurol. Neurosurg. 2022, 214, 107127. [Google Scholar] [CrossRef]
- Merimsky, O.; Lepechoux, C.; Terrier, P.; Vanel, D.; Delord, J.P.; LeCesne, A. Primary Sarcomas of the Central Nervous System. Oncology 2000, 58, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.; Kaste, S.; Jenkins, J.; Hewan-Lowe, K.; Spence, J.; Gupta, M.; Monson, D.; Fletcher, B. Epithelioid Sarcoma: Clinical, MR Imaging and Pathologic Findings. Skelet. Radiol. 2002, 31, 400–412. [Google Scholar] [CrossRef]
- Gounder, M.M.; Merriam, P.; Ratan, R.; Patel, S.R.; Chugh, R.; Villalobos, V.M.; Thornton, M.; Van Tine, B.A.; Abdelhamid, A.H.; Whalen, J.; et al. Real-world Outcomes of Patients with Locally Advanced or Metastatic Epithelioid Sarcoma. Cancer 2021, 127, 1311–1317. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.L.; Ong, W.; Tan, J.H.; Kumar, N.; Hallinan, J.T.P.D. Epithelioid Sarcoma of the Spine: A Review of Literature and Case Report. J. Clin. Med. 2023, 12, 5632. https://doi.org/10.3390/jcm12175632
Tan YL, Ong W, Tan JH, Kumar N, Hallinan JTPD. Epithelioid Sarcoma of the Spine: A Review of Literature and Case Report. Journal of Clinical Medicine. 2023; 12(17):5632. https://doi.org/10.3390/jcm12175632
Chicago/Turabian StyleTan, Yi Liang, Wilson Ong, Jiong Hao Tan, Naresh Kumar, and James Thomas Patrick Decourcy Hallinan. 2023. "Epithelioid Sarcoma of the Spine: A Review of Literature and Case Report" Journal of Clinical Medicine 12, no. 17: 5632. https://doi.org/10.3390/jcm12175632
APA StyleTan, Y. L., Ong, W., Tan, J. H., Kumar, N., & Hallinan, J. T. P. D. (2023). Epithelioid Sarcoma of the Spine: A Review of Literature and Case Report. Journal of Clinical Medicine, 12(17), 5632. https://doi.org/10.3390/jcm12175632