
Primary Hemostasis Disorders as a Cause of Heavy Menstrual Bleeding in Women of Reproductive Age

 ,
,  , ,
, , 

Abstract
:
1. Introduction
2. Primary Hemostasis Disorders
2.1. Von Willebrand Disease
2.1.1. Pathophysiology
2.1.2. Classification
2.1.3. Inheritance Pattern
2.1.4. Clinical Manifestations and Epidemiology of HMB
2.1.5. Diagnosis
- Heavy menstrual bleeding since menarche.
- One of the following conditions:
- ■
- Postpartum hemorrhage,
- ■
- Surgery-related bleeding, and
- ■
- Bleeding associated with dental work.
- Two or more of the following conditions:
- ■
- Epistaxis, one to two times per month,
- ■
- Frequent gum bleeding, and
- ■
- Family history of bleeding symptoms.
2.1.6. Treatment Options for HMB
2.2. Glanzmann Thrombasthenia
2.2.1. Pathophysiology
2.2.2. Clinical Manifestations
2.2.3. Epidemiology of HMB
2.2.4. Diagnosis
2.2.5. Treatment Options
- (1)
- Preventative measures, such as abstinence from bleeding triggers (such as NSAIDS);
- (2)
- Topical measures, such as packing in case of epistaxis;
- (3)
- Antifibrinolytics, mainly used in surgeries and HMB;
- (4)
- DDAVP;
- (5)
- Recombinant Factor VIIa (rFVIIa);
- (6)
- Female hormones for gynecologic complications;
- (7)
- Surgical interventions (such as dilatation and curettage);
- (8)
- Red Blood Cell transfusions;
- (9)
- Platelet transfusions [43]. Platelet transfusions are considered standard treatment during surgeries or trauma and are often used in nonsurgical episodes [43,44]. However, platelet alloimmunization as well as blood borne infection transmission should be taken into consideration every time this therapeutic intervention is considered [45].
2.2.6. Treatment Options for HMB
- (A)
- (B)
- (C)
- LNG-IUS [49].
2.3. Bernard–Soulier Syndrome
2.3.1. Pathophysiology
2.3.2. Clinical Manifestations
2.3.3. Epidemiology of HMB
2.3.4. Diagnosis
2.3.5. Treatment Options for HMB
2.4. Hermansky–Pudlak Syndrome
2.4.1. Pathophysiology
2.4.2. Clinical Manifestations
2.4.3. Epidemiology of HMB
2.4.4. Diagnosis
2.4.5. Treatment Options for HMB
2.5. Immune Thrombocytopenia
2.5.1. Pathophysiology
2.5.2. Clinical Manifestations
2.5.3. Epidemiology
2.5.4. Diagnosis
2.5.5. Treatment Options
2.5.6. Treatment Options for HMB
2.6. Joint Hypermobility
2.6.1. Definitions and Clinical Manifestations
2.6.2. Epidemiology
2.6.3. Epidemiology of HMB
2.6.4. Pathophysiology
2.6.5. Treatment Options for HMB
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagberg, K.W.; Jick, S.; Du, P.; Berthoz, F.T.; Özen, G.; Tzivelekis, S. Impact of von Willebrand Disease on Women’s Health Outcomes: A Matched Cohort Database Study. J. Women’s Health 2022, 31, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Rajpurkar, M. Challenges of diagnosing and managing the adolescent with heavy menstrual bleeding. Thromb. Res. 2016, 143, 91–100. [Google Scholar] [CrossRef]
- Holm, E.; Carlsson, K.S.; Lövdahl, S.; Lail, A.E.; Abshire, T.C.; Berntorp, E. Bleeding-related hospitalization in patients with von Willebrand disease and the impact of prophylaxis: Results from national registers in Sweden compared with normal controls and participants in the von Willebrand Disease Prophylaxis Network. Haemophilia 2018, 24, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Brighton, S.; Laffan, M.; Goudemand, J.; Franks, B.; Finnegan, A. The Cost of Von Willebrand Disease in Europe: The CVESS Study. Clin. Appl. Thromb. 2022, 28, 10760296221120583. [Google Scholar] [CrossRef]
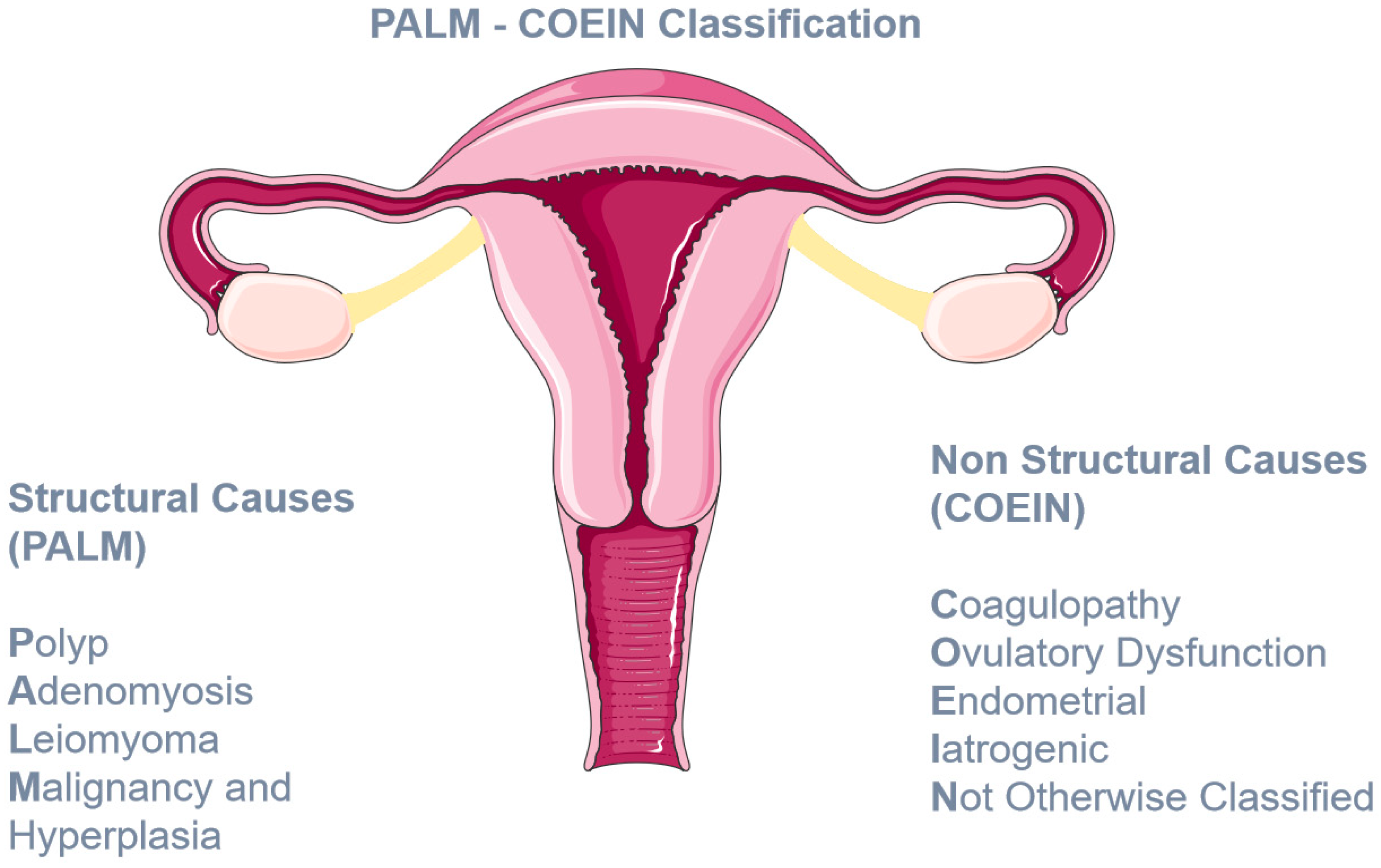
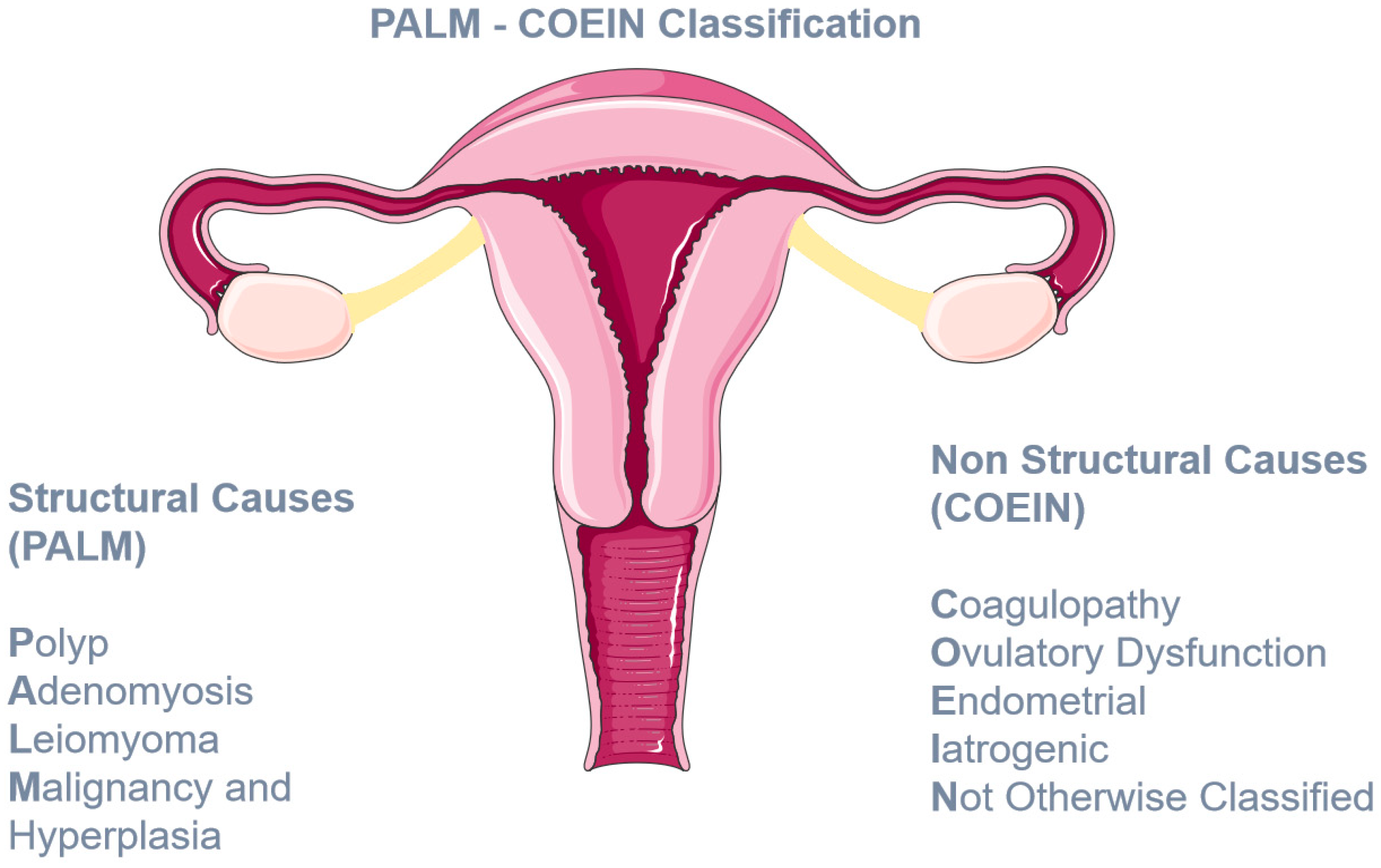
- Munro, M.G.; Critchley, H.O.; Broder, M.S.; Fraser, I.S.; for the FIGO Working Group on Menstrual Disorders. FIGO classification system (PALM-COEIN) for causes of abnormal uterine bleeding in nongravid women of reproductive age. Int. J. Gynecol. Obstet. 2011, 113, 3–13. [Google Scholar] [CrossRef]
- Van Ommen, C.H.; Peters, M. Clinical practice: The bleeding child. Part I: Primary hemostatic disorders. Eur. J. Pediatr. 2012, 171, 1–10. [Google Scholar] [PubMed]
- Nurden, A.T. Glanzmann thrombasthenia. Orphanet J. Rare Dis. 2006, 1, 10. [Google Scholar] [CrossRef]
- Goodeve, A.; James, P. Von Willebrand Disease. In GeneReviews® [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2009. [Google Scholar]
- López, J.A.; Andrews, R.K.; Afshar-Kharghan, V.; Berndt, M.C. Bernard-Soulier syndrome. Blood 1998, 91, 4397–4418. [Google Scholar] [CrossRef]
- Gahl, W.A.; Brantly, M.; Kaiser-Kupfer, M.I.; Iwata, F.; Hazelwood, S.; Shotelersuk, V.; Duffy, L.F.; Kuehl, E.M.; Troendle, J.; Bernardini, I. Genetic Defects and Clinical Characteristics of Patients with a Form of Oculocutaneous Albinism (Hermansky–Pudlak Syndrome). New. Engl. J. Med. 1998, 338, 1258–1265. [Google Scholar] [CrossRef]
- Yew, K.S.; Kamps-Schmitt, K.A.; Borge, R. Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders. Am. Fam. Physician 2021, 103, 481–492. [Google Scholar]
- Anonymous. ACOG Committee Opinion No. 451: Von Willebrand Disease in Women. Obstet. Gynecol. 2009, 114, 1439–1443. [Google Scholar] [CrossRef]
- Shankar, M.; Lee, C.A.; Sabin, C.A.; Economides, D.L.; Kadir, R.A. von Willebrand disease in women with menorrhagia: A systematic review. BJOG: Int. J. Obstet. Gynaecol. 2004, 111, 734–740. [Google Scholar] [CrossRef] [PubMed]
- James, A.H. Obstetric Management of Adolescents with Bleeding Disorders. J. Pediatr. Adolesc. Gynecol. 2010, 23, S31–S37. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E.; Budde, U.; Eikenboom, J.C.; Favaloro, E.J.; Hill, F.G.; Holmberg, L.; Ingerslev, J.; Lee, C.A.; Lillicrap, D.; Mannucci, P.M.; et al. Update on the pathophysiology and classification of von Willebrand disease: A report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 2006, 4, 2103–2114. [Google Scholar] [CrossRef]
- Stockschlaeder, M.; Schneppenheim, R.; Budde, U. Update on von Willebrand factor multimers. Blood Coagul. Fibrinolysis 2014, 25, 206–216. [Google Scholar] [CrossRef]
- Leebeek, F.W.G.; Eikenboom, J.C.J. Von Willebrand’s Disease. New. Engl. J. Med. 2016, 375, 2067–2080. [Google Scholar] [CrossRef] [PubMed]
- Von Willebrand, E.A. Hereditary pseudohemophilia. Fin. Lakaresallsk. Handl. 1926, 68, 87–112. [Google Scholar]
- James, P.D.; Goodeve, A.C. Von Willebrand disease. Genet. Med. 2011, 13, 365–376. [Google Scholar] [CrossRef]
- Boisseau, P.; Giraud, M.; Ternisien, C.; Veyradier, A.; Fressinaud, E.; Lefrancois, A.; Bezieau, S.; Fouassier, M. An unexpected transmission of von Willebrand disease type 3: The first case of maternal uniparental disomy 12. Haematologica 2011, 96, 1567–1568. [Google Scholar] [CrossRef]
- Weyand, A.C.; Flood, V.H. Von Willebrand Disease. Hematol. Oncol. Clin. N. Am. 2021, 35, 1085–1101. [Google Scholar] [CrossRef]
- Veyradier, A.; Boisseau, P.; Fressinaud, E.; Caron, C.; Ternisien, C.; Giraud, M.; Zawadzki, C.; Trossaert, M.; Itzhar-Baïkian, N.; Dreyfus, M.; et al. A Laboratory Phenotype/Genotype Correlation of 1167 French Patients From 670 Families With von Willebrand Disease. Medicine 2016, 95, e3038. [Google Scholar] [CrossRef]
- Ragni, M.V.; Machin, N.; Malec, L.M.; James, A.H.; Kessler, C.M.; Konkle, B.A.; Kouides, P.A.; Neff, A.T.; Philipp, C.S.; Brambilla, D.J. Von Willebrand factor for menorrhagia: A survey and literature review. Haemophilia 2016, 22, 397–402. [Google Scholar] [CrossRef]
- Sanders, Y.V.; De Wee, E.M.; Meijer, K.; Eikenboom, J.; Van Der Bom, J.G.; Fijnvandraat, C.J.K.; Gorkom, B.A.P.L.-V.; Cnossen, M.H.; Mauser-Bunschoten, E.P.; Leebeek, F.W.G. Von Willebrand disease in the Netherlands: The WiN study. Ned. Tijdschr. voor Geneeskd. 2014, 158, A6518. [Google Scholar]
- De Wee, E.M.; Knol, H.M.; Mauser-Bunschoten, E.P.; van der Bom, J.G.; Eikenboom, J.C.; Fijnvandraat, K.; De Goede-Bolder, A.; Laros-van Gorkom, B.; Ypma, P.F.; Zweegman, S.; et al. Gynaecological and obstetric bleeding in moderate and severe von Willebrand disease. Thromb. Haemost. 2011, 106, 885–892. [Google Scholar] [CrossRef]
- Kadir, R.A.; Economides, D.L.; Sabin, C.A.; Owens, D.; Lee, C.A. Frequency of inherited bleeding disorders in women with menor-rhagia. Lancet 1998, 351, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.I.; Meschengieser, S.S.; Blanco, A.N.; Salviú, M.J.; Farías, E.C.; Kempfer, A.C.; Lazzari, A.M. Clinical features and laboratory patterns in a cohort of consecutive Argentinian patients with von Willebrand’s disease. Haematologica 2001, 86, 420–427. [Google Scholar]
- James, P.D.; Connell, N.T.; Ameer, B.; Di Paola, J.; Eikenboom, J.; Giraud, N.; Haberichter, S.; Jacobs-Pratt, V.; Konkle, B.; McLintock, C.; et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021, 5, 280–300. [Google Scholar] [CrossRef]
- Brignardello-Petersen, R.; El Alayli, A.; Husainat, N.; Kalot, M.A.; Shahid, S.; Aljabirii, Y.; Britt, A.; Alturkmani, H.J.; El-Khechen, H.; Motaghi, S.; et al. Gynecologic and obstetric management of women with von Willebrand disease: Summary of 3 systematic reviews of the literature. Blood Adv. 2022, 6, 228–237. [Google Scholar] [CrossRef]
- Kouides, P.A.; Conard, J.; Peyvandi, F.; Lukes, A.; Kadir, R. Hemostasis and menstruation: Appropriate investigation for underlying disorders of hemostasis in women with excessive menstrual bleeding. Fertil. Steril. 2005, 84, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Kouides, P.A.; Byams, V.R.; Philipp, C.S.; Stein, S.F.; Heit, J.A.; Lukes, A.S.; Skerrette, N.I.; Dowling, N.F.; Evatt, B.L.; Miller, C.H.; et al. Multisite management study of menorrhagia with abnormal laboratory haemostasis: A prospective crossover study of intranasal desmopressin and oral tranexamic acid. Br. J. Haematol. 2009, 145, 212–220. [Google Scholar] [CrossRef]
- Amesse, L.S.; Pfaff-Amesse, T.; Leonardi, R.; Uddin, D.; French, J.A. Oral Contraceptives and DDAVP Nasal Spray: Patterns of Use in Managing vWD-Associated Menorrhagia. J. Pediatr. Hematol. Oncol. 2005, 27, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Ragni, M.V.; Rothenberger, S.D.; Feldman, R.; Nance, D.; Leavitt, A.D.; Malec, L.; Kulkarni, R.; Sidonio, R.; Kraut, E.; Lasky, J.; et al. Recombinant von Willebrand factor and tranexamic acid for heavy menstrual bleeding in patients with mild and moderate von Willebrand disease in the USA (VWDMin): A phase 3, open-label, randomised, crossover trial. Lancet Haematol. 2023, 10, e612–e623. [Google Scholar] [CrossRef] [PubMed]
- Connell, N.T.; Flood, V.H.; Brignardello-Petersen, R.; Abdul-Kadir, R.; Arapshian, A.; Couper, S.; Grow, J.M.; Kouides, P.; Laffan, M.; Lavin, M.; et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021, 5, 301–325. [Google Scholar] [CrossRef]
- Solh, M.; Solh, T.; Botsford, A. Glanzmann’s thrombasthenia: Pathogenesis, diagnosis, and current and emerging treatment options. J. Blood Med. 2015, 6, 219–227. [Google Scholar] [CrossRef]
- Toogeh, G.; Sharifian, R.; Lak, M.; Safaee, R.; Artoni, A.; Peyvandi, F. Presentation and pattern of symptoms in 382 patients with Glanzmann thrombasthenia in Iran. Am. J. Hematol. 2004, 77, 198–199. [Google Scholar] [CrossRef]
- George, J.; Caen, J.; Nurden, A. Glanzmann’s thrombasthenia: The spectrum of clinical disease. Blood 1990, 75, 1383–1395. [Google Scholar] [CrossRef]
- Di Minno, G.; Zotz, R.B.; d’Oiron, R.; Bindslev, N.; Di Minno, M.N.; Poon, M.C.; Glanzmann Thrombasthenia Registry Investigators. The international prospective Glanzmann Throm-basthenia Registry: Treatment modalities and outcomes in non-surgical bleeding episodes in Glanzmann thrombasthenia pa-tients. Haematologica 2015, 100, 1031–1037. [Google Scholar]
- Poon, M.C.; d’Oiron, R.; Zotz, R.B.; Bindslev, N.; Di Minno, M.N.; Di Minno, G.; Glanzmann Thrombasthenia Registry Investigators. The international prospective Glanzmann Throm-basthenia Registry: Treatment and outcomes in surgical intervention. Haematologica 2015, 100, 1038–1044. [Google Scholar]
- Franchini, M.; Favaloro, E.J.; Lippi, G. Glanzmann thrombasthenia: An update. Clin. Chim. Acta 2010, 411, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Punt, M.C.; Schuitema, P.C.E.; Bloemenkamp, K.W.M.; Hovinga, I.C.L.K.; Galen, K.P.M. Menstrual and obstetrical bleeding in women with inherited platelet receptor defects—A systematic review. Haemophilia 2020, 26, 216–227. [Google Scholar] [CrossRef]
- Grainger, J.D.; Thachil, J.; Will, A.M. How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. Br. J. Haematol. 2018, 182, 621–632. [Google Scholar] [CrossRef]
- Seligsohn, U. Treatment of inherited platelet disorders. Haemophilia 2012, 18, 161–165. [Google Scholar] [CrossRef] [PubMed]
- King, L.J.; Huff, J.; Heber, D.; Miller, M.A.; Marshall, B. Management of Refractory Menstrual Bleeding in an Adolescent with Glanzmann Thrombasthenia: A Case Report and Review. Case Rep. Obstet. Gynecol. 2020, 2020, 1–3. [Google Scholar] [CrossRef]
- Poon, M.C.; Di Minno, G.; d’Oiron, R.; Zotz, R. New Insights into the Treatment of Glanzmann Thrombasthenia. Transfus. Med. Rev. 2016, 30, 92–99. [Google Scholar] [CrossRef]
- Markovitch, O.; Ellis, M.; Holzinger, M.; Goldberger, S.; Beyth, Y. Severe juvenile vaginal bleeding due to Glanzmann’s thrombas-thenia: Case report and review of the literature. Am. J. Hematol. 1998, 57, 225–227. [Google Scholar] [CrossRef]
- Ezenwosu, O.U.; Chukwu, B.F.; Uwaezuoke, N.A.; Ezenwosu, I.L.; Ikefuna, A.N.; Emodi, J.I. Glanzmann’s thrombasthenia: A rare bleeding disorder in a Nigerian girl. Afr. Health Sci. 2020, 20, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Balci, Y.I.; Karabulut, A.; Kabukcu, S.; Sari, I.; Keskin, A. Intensive Menstrual Bleeding Successfully Treated with Recombinant Factor VIIa in Glanzmann Thrombasthenia. Clin. Appl. Thromb. 2011, 17, 320–322. [Google Scholar] [CrossRef]
- Lu, M.; Yang, X. Levonorgestrel-releasing intrauterine system for treatment of heavy menstrual bleeding in adolescents with Glanzmann’s Thrombasthenia: Illustrated case series. BMC Women’s Health 2018, 18, 1–5. [Google Scholar] [CrossRef]
- Rajpurkar, M.; O’Brien, S.H.; Haamid, F.W.; Cooper, D.L.; Gunawardena, S.; Chitlur, M. Heavy Menstrual Bleeding as a Common Presenting Symptom of Rare Platelet Disorders: Illustrative Case Examples. J. Pediatr. Adolesc. Gynecol. 2016, 29, 537–541. [Google Scholar] [CrossRef]
- Jimenez, J.S.; Martin, I.; de La Fuente, L.; Mu-Oz, J.L.; Vaquero, G.; Ramirez, M.; Perez, C.; de La Fuente, P. Severe menorrhagia due to Glanzmann throm-basthenia treated with hydrothermal ablation. J. Am. Assoc. Gynecol. Laparosc. 2000, 7, 265–267. [Google Scholar] [CrossRef]
- Kushwaha, R. Haemostatic Disorder in Women with Unexplained Menorrhagia: A Tertiary Care Centre Experience from Northern India. J. Clin. Diagn. Res. 2017, 11, EC46. [Google Scholar] [CrossRef]
- Simon, D.; Kunicki, T.; Nugent, D. Platelet function defects. Haemophilia 2008, 14, 1240–1249. [Google Scholar] [CrossRef]
- Warad, D.; Chitlur, M.; Philipp, C. Platelet Disorders in the Adolescent Female. In Hematology in the Adolescent Female; Springer International Publishing: Cham, Switzerland, 2020; pp. 61–78. [Google Scholar]
- Obeng-Tuudah, D.; Hussein, B.A.; Hakim, A.; Gomez, K.; Kadir, R.A. The presentation and outcomes of Hermansky-Pudlak syndrome in obstetrics and gynecological settings: A systematic review. Int. J. Gynecol. Obstet. 2021, 154, 412–426. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Darnige, L.; Bellucci, S. Molecular determinants of platelet delta storage pool deficiencies: An update. Br. J. Haematol. 2013, 160, 5–11. [Google Scholar] [CrossRef]
- Introne, W.J.; Huizing, M.; Malicdan, M.C.V.; O’Brien, K.J.; Gahl, W.A. Hermansky-Pudlak Syndrome. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lohse, J.; Gehrisch, S.; Tauer, J.T.; Knöfler, R. Therapy refractory menorrhagia as first manifestation of Hermansky-Pudlak syndrome. Hamostaseologie 2011, 31 (Suppl. 1), S61–S63. [Google Scholar]
- Ray, A. Case Report: Hermansky Pudlak Syndrome (Presenting as late onset heavy Menstrual Bleeding). J. Clin. Diagn. Res. 2013, 7, 2023–2024. [Google Scholar] [CrossRef] [PubMed]
- Kingman, C.; Kadir, R.; Lee, C.; Economides, D. The use of levonorgestrel-releasing intrauterine system for treatment of menorrhagia in women with inherited bleeding disorders. BJOG Int. J. Obstet. Gynaecol. 2004, 111, 1425–1428. [Google Scholar] [CrossRef]
- Rivera-Concepción, J.; Acevedo-Canabal, J.; Burés, A.; Vargas, G.; Cadilla, C.; Izquierdo, N.J. Bleeding assessment in female patients with the Hermansky-Pudlak syndrome—A case series. Eur. J. Haematol. 2019, 102, 432–436. [Google Scholar] [CrossRef]
- Cooper, N.; Kruse, A.; Kruse, C.; Watson, S.; Morgan, M.; Provan, D.; Ghanima, W.; Arnold, D.M.; Tomiyama, Y.; Santoro, C.; et al. Immune thrombocytopenia (ITP) World Impact Survey (iWISh): Patient and physician perceptions of diagnosis, signs and symptoms, and treatment. Am. J. Hematol. 2021, 96, 188–198. [Google Scholar] [CrossRef]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Marranconi, E. Management of immune thrombocytopenia in women: Current standards and special considerations. Expert Rev. Hematol. 2020, 13, 175–185. [Google Scholar] [CrossRef]
- Andrès, E.; Mecili, M.; Fothergill, H.; Zimmer, J.; Vogel, T.; Maloisel, F. Gender-related analysis of the clinical presentation, treatment response and outcome in patients with immune thrombocytopenia. La Presse Médicale 2012, 41, e426–e431. [Google Scholar] [CrossRef]
- Cines, D.B.; Blanchette, V.S. Immune Thrombocytopenic Purpura. New Engl. J. Med. 2002, 346, 995–1008. [Google Scholar] [CrossRef]
- Kistangari, G.; McCrae, K.R. Immune Thrombocytopenia. Hematol. Oncol. Clin. N. Am. 2013, 27, 495–520. [Google Scholar] [CrossRef]
- Efficace, F.; Mandelli, F.; Fazi, P.; Santoro, C.; Gaidano, G.; Cottone, F.; Borchiellini, A.; Carpenedo, M.; Simula, M.P.; Di Giacomo, V.; et al. Health-related quality of life and burden of fatigue in patients with primary immune thrombocytopenia by phase of disease. Am. J. Hematol. 2016, 91, 995–1001. [Google Scholar] [CrossRef]
- Van Dijk, W.E.M.; Punt, M.C.; van Galen, K.P.M.; van Leeuwen, J.; Lely, A.T.; Schutgens, R.E.G. Menstrual problems in chronic immune thrombocytopenia: A monthly challenge—A cohort study and review. Br. J. Haematol. 2022, 198, 753–764. [Google Scholar] [CrossRef]
- NICE. Heavy Menstrual Bleeding: Assessment and Management (NICE Guideline, No. 88.); National Institute for Health and Care Excellence: London, UK, 2021. [Google Scholar]
- James, A.H. Heavy menstrual bleeding: Work-up and management. Hematology 2016, 2016, 236–242. [Google Scholar] [CrossRef]
- Malek, S.; Reinhold, E.J.; Pearce, G.S. The Beighton Score as a measure of generalised joint hypermobility. Rheumatol. Int. 2021, 41, 1707–1716. [Google Scholar] [CrossRef]
- Kendel, N.E.; Haamid, F.W.; Christian-Rancy, M.; O’Brien, S.H. Characterizing adolescents with heavy menstrual bleeding and generalized joint hypermobility. Pediatr. Blood Cancer 2019, 66, e27675. [Google Scholar] [CrossRef]
- Fikree, A.; Aziz, Q.; Grahame, R. Joint Hypermobility Syndrome. Rheum. Dis. Clin. N. Am. 2013, 39, 419–430. [Google Scholar] [CrossRef]
- Moyer, G.; Huguelet, P.; Trapane, P. Hypermobility Syndromes in Heavy Menstrual Bleeding. In Hematology in the Adolescent Female; Springer International Publishing: Cham, Switzerland, 2020; pp. 89–98. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Jesudas, R.; Chaudhury, A.; Laukaitis, C.M. An update on the new classification of Ehlers-Danlos syndrome and review of the causes of bleeding in this population. Haemophilia 2019, 25, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Al-Rawi, Z.S.; Al-Aszawi, A.J.; Al-Chalabi, T. Joint Mobility Among University Students in Iraq. Rheumatology 1985, 24, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Gocentas, A.; Jascaniniene, N.; Pasek, M.; Przybylski, W.; Matulyte, E.; Mieliauskaite, D.; Kwilecki, K.; Jaszczanin, J. Prevalence of generalised joint hy-permobility in school-aged children from east-central European region. Folia. Morphol. 2016, 75, 48–52. [Google Scholar] [CrossRef]
- Demmler, J.C.; Atkinson, M.D.; Reinhold, E.J.; Choy, E.; Lyons, A.R.; Brophy, S.T. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: A national electronic cohort study and case–control comparison. BMJ Open 2019, 9, e031365. [Google Scholar] [CrossRef]
- Hugon-Rodin, J.; Lebègue, G.; Becourt, S.; Hamonet, C.; Gompel, A. Gynecologic symptoms and the influence on reproductive life in 386 women with hypermobility type Ehlers-Danlos Syndrome: A Cohort Study. Orphanet J. Rare Dis. 2016, 11, 1–6. [Google Scholar] [CrossRef]
- Castori, M.; Morlino, S.; Dordoni, C.; Celletti, C.; Camerota, F.; Ritelli, M.; Morrone, A.; Venturini, M.; Grammatico, P.; Colombi, M. Gynecologic and obstetric implications of the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome hypermobility type) in 82 Italian patients. Am. J. Med. Genet. Part A 2012, 158A, 2176–2182. [Google Scholar] [CrossRef]
- Kumskova, M.; Flora, G.D.; Staber, J.; Lentz, S.R.; Chauhan, A.K. Characterization of bleeding symptoms in Ehlers–Danlos syndrome. J. Thromb. Haemost. 2023, 21, 1824–1830. [Google Scholar] [CrossRef]
- Artoni, A.; Bassotti, A.; Abbattista, M.; Marinelli, B.; Lecchi, A.; Gianniello, F.; Clerici, M.; Bucciarelli, P.; La Marca, S.; Peyvandi, F.; et al. Hemostatic abnormalities in patients with Ehlers–Danlos syndrome. J. Thromb. Haemost. 2018, 16, 2425–2431. [Google Scholar] [CrossRef]
- Hernandez, A.M.C.; Dietrich, J.E. Gynecologic Management of Pediatric and Adolescent Patients with Ehlers-Danlos Syndrome. J. Pediatr. Adolesc. Gynecol. 2020, 33, 291–295. [Google Scholar] [CrossRef]
- Huguelet, P.S.; Laurin, J.; Thornhill, D.; Moyer, G. Use of the Levonorgestrel Intrauterine System to Treat Heavy Menstrual Bleeding in Adolescents and Young Adults with Inherited Bleeding Disorders and Ehlers-Danlos Syndrome. J. Pediatr. Adolesc. Gynecol. 2021, 35, 147–152.e1. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study Population | Prevalence of HMB | |
|---|---|---|
| Ragni et al., 2016 [23] | 1321 women with VWD from 20 US Hemophilia Treatment Centers, 18–45 years old, seen during 2012–2014. | Heavy menstrual bleeding reported by 816 (61.8%) women with VWD. |
| Sanders et al., 2014 [24] | 664 adults with Von Willebrand disease, as compared with 500 healthy persons, in the Willebrand in the Netherlands (WiN) study. | More than 80% of women with VWD experienced menorrhagia. |
| de Wee et al., 2011 [25] | 423 women aged ≥ 16 years old with moderate and severe VWD in the Netherlands. | Menorrhagia, defined as occurrence of ≥2 menorrhagia symptoms, was reported by 81%. |
| Kadir et al., 1998 [26] | 150 women referred for investigation of menorrhagia whose pelvis was normal on clinical examination and who had an estimated menstrual blood loss of more than 80 mL. | 13% VWD prevalence. Menorrhagia since menarche 65% of 20 women with Von Willebrand disease compared with 8,9% of 123 women without a bleeding disorder. |
| Woods et al., 2001 [27] | 1885 patients of all ages with VWD–1142 females—from a reference center in Argentina. | 47% of women more than 13 years old. |
| Therapeutic Interventions | Source of Evidence | Study Population | Results |
|---|---|---|---|
| Desmopressin vs. TxA | 1 Randomized clinical trial [31] | 116 women 18–50 years old with HMB and coagulation disorder. | (1) Average estimated decrease in PBAC * score from baseline: −64·1 (95% CI = −88·0, −40·3) for IN-DDAVP and −105·7 (95% CI = −130·5, −81·0) for TxA. (2) Normalization of menstrual bleeding (PBAC < 100): 22% for IN-DDAVP vs. 33% for TxA. |
| Desmopressin vs. hormonal therapy | 1 Observational study [32] | 36 adolescent females 9–18 years old with HMB and VWD. | Alleviation of symptoms (median follow-up 30 months): 77.1% DDAVP vs. 85.7% hormonal therapy RR 0.90; 95% CI, 0.66–1.23). |
| TxA vs. recombinant VWF | 1 Randomized cross-over clinical trial [33] | 36 women 13–45 years old with mild or moderate VWF and HMB. | Median PBAC score significantly lower after two cycles with tranexamic acid vs. recombinant VWF (146 (95% CI 117–199) vs. 213 (152–298); adjusted mean treatment difference 46 (95% CI 2–90); p = 0·039). (median follow-up 23.97 weeks). |
| Number of Patients | Treatment Method |
|---|---|
| 16/18 (88.9%) | Hormonal Methods |
| 15/18 (83.3%) | Blood products |
| 9/18 (50%) | Antifibrinolytics |
| 4/18 (22.2%) | rFVIIa |
| 4/18 (22.2%) | Surgical interventions |
| 3/18 (16.7%) | Iron Supplementation |
| Number of Patients | Treatment Method |
|---|---|
| 13/14 (92.9%) | Blood products |
| 11/14 (78.6%) | Hormonal methods |
| 9/14 (64.3%) | Iron supplementation |
| 8/14 (57.1%) | Antifibrinolytics |
| 3/14 (21.4%) | rFVIIa |
| 3/14 (21.4%) | Surgery |
| 2/14 (14.3%) | DDAVP |
| 3/14 (21.4%) | Other (uterotonic agents, steroids and IVIG) |
| Publication | Patient | Treatment |
|---|---|---|
| J Lohse et al. [58] | 13-year-old with 14 days HMB at menarche that led to diagnosis of HPS | 8 PRBCs, 1 FFP and Norethisterone could not control the bleeding |
| After suspicion of the diagnosis 5 doses of IV desmopressin and IV tranexamic acid were also not effective. Then, rFVIIa was used and bleeding was controlled. She continued during her periods with progesterone and TXA for further prevention in the long run. | ||
| Ray A et al. [59] | 42-year-old with 7-month history of HMB that led to diagnosis of HPS | PRBCs (unknown number), Oral contraceptives, TxA and Desmopressin that controlled her bleeding. She continued with TxA and desmopressin during her periods. |
| Number of Patients | Treatment Method |
|---|---|
| 4/6 (66.7%) | Hormonal therapy |
| 2/6 (33.3%) | Aminocaproic acid |
| 2/6 (33.3%) | Desmopressin |
| 2/6 (33.3%) | Surgical intervention (1 hysterectomy and I endometrial ablation) |
| 1/6 (16.7%) | Platelet transfusion |
| 1/6 (16.7%) | Iron supplementation |
| Management Method | Number of Patients |
|---|---|
| OCP monotherapy | 7 (19%) |
| LNG-IUD monotherapy | 10 (27%) |
| Hormonal therapy, other | 2 (5%) |
| OCP + TxA | 2 (5%) |
| LNG-ICP + TxA | 1 (3%) |
| TxA + IVIG | 1 (3%) |
| Passive Hyperextension of the Fifth Metacarpophalangeal Joint beyond 90° | 1 Point for Each Side of the Body (Left, Right) |
|---|---|
| Passive apposition of the thumb to the forearm | 1 point for each side |
| Hyperextension of the elbow beyond 10° | 1 point for each side |
| Hyperextension of the knee beyond 10° | 1 point for each side |
| Placing of palms flat on the floor while flexing forward the spine with knees extended | 1 point |
| Sum/9 |
| Hypermobility Type Diagnosis | Beighton Score | Musculoskeletal Manifestations * | |
|---|---|---|---|
| Hypermobile Ehlers–Danlos Syndrome | Positive | Possible | Meets diagnostic criteria for hEDS |
| Hypermobility Spectrum Disorders (HSD) | |||
| Generalized HSD | Positive | Present | Does not meet criteria for hEDS |
| Peripheral HSD | Usually negative | Present | JH limited to hands/feet |
| Localized HSD | Negative | Present | JH limited to certain joints or body parts |
| Historical HSD | Negative | Present | Reported historic presence of JH |
| Asymptomatic non-Syndromic JH | |||
| Asymptomatic generalized JH | Positive | Absent | |
| Asymptomatic peripheral JH | Usually negative | Absent | JH limited to hands/feet |
| Asymptomatic localized JH | Negative | Absent | JH limited to certain joints or body part (usually less than 5 joints) |
| Diagnosis of hEDS Requires Simultaneous Presence of Criteria 1, 2 and 3 |
|---|
| Criterion 1: Generalized joint hypermobility (GJH) as indicated by the Beighton score (a) ≥4/9 for those >50 years of age (b) ≥5/9 for pubertal men and women up to the age of 50 (c) ≥6/9 for pre-pubertal children and adolescents |
| Criterion 2: 2 of 3 must be met (a) ≥5/12 systemic findings in skin, heart, etc. (b) Family history of hEDS (c) Joint pain or instability |
| Criterion 3: Absence of other causes of symptoms (a) Absence of other types of EDS (b) Exclusion of other connective tissue disorders (c) Exclusion of other causes of joint hypermobility |
| Number of Patients | Treatment Method |
|---|---|
| 19/30 (63%) | Long-term oral hormonal therapy |
| 3/30 (10%) | Oral hormonal therapy + intrauterine device (IUD) |
| 3/30 (10%) | DMPA (Depo Medroxyprogesterone Acetate) |
| 2/30 (6.6%) | IUD |
| 2/30 (6.6%) | Tranexamic acid |
| 1/30 (3.3%) | Oral hormonal therapy +IUD +leuprolide acetate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kontogiannis, A.; Matsas, A.; Valsami, S.; Livanou, M.E.; Panoskaltsis, T.; Christopoulos, P. Primary Hemostasis Disorders as a Cause of Heavy Menstrual Bleeding in Women of Reproductive Age. J. Clin. Med. 2023, 12, 5702. https://doi.org/10.3390/jcm12175702
Kontogiannis A, Matsas A, Valsami S, Livanou ME, Panoskaltsis T, Christopoulos P. Primary Hemostasis Disorders as a Cause of Heavy Menstrual Bleeding in Women of Reproductive Age. Journal of Clinical Medicine. 2023; 12(17):5702. https://doi.org/10.3390/jcm12175702
Chicago/Turabian StyleKontogiannis, Athanasios, Alkis Matsas, Serena Valsami, Maria Effrosyni Livanou, Theodoros Panoskaltsis, and Panagiotis Christopoulos. 2023. "Primary Hemostasis Disorders as a Cause of Heavy Menstrual Bleeding in Women of Reproductive Age" Journal of Clinical Medicine 12, no. 17: 5702. https://doi.org/10.3390/jcm12175702
APA StyleKontogiannis, A., Matsas, A., Valsami, S., Livanou, M. E., Panoskaltsis, T., & Christopoulos, P. (2023). Primary Hemostasis Disorders as a Cause of Heavy Menstrual Bleeding in Women of Reproductive Age. Journal of Clinical Medicine, 12(17), 5702. https://doi.org/10.3390/jcm12175702