

B Cell Tolerance and Targeted Therapies in SLE



Abstract
:1. Introduction and Scope
2. B Cell Development and Subpopulations
3. Central and Peripheral B Cell Tolerance
4. Regulation of B Cells by Innate Immune Cells
5. B Cell Functions in Autoimmunity and SLE
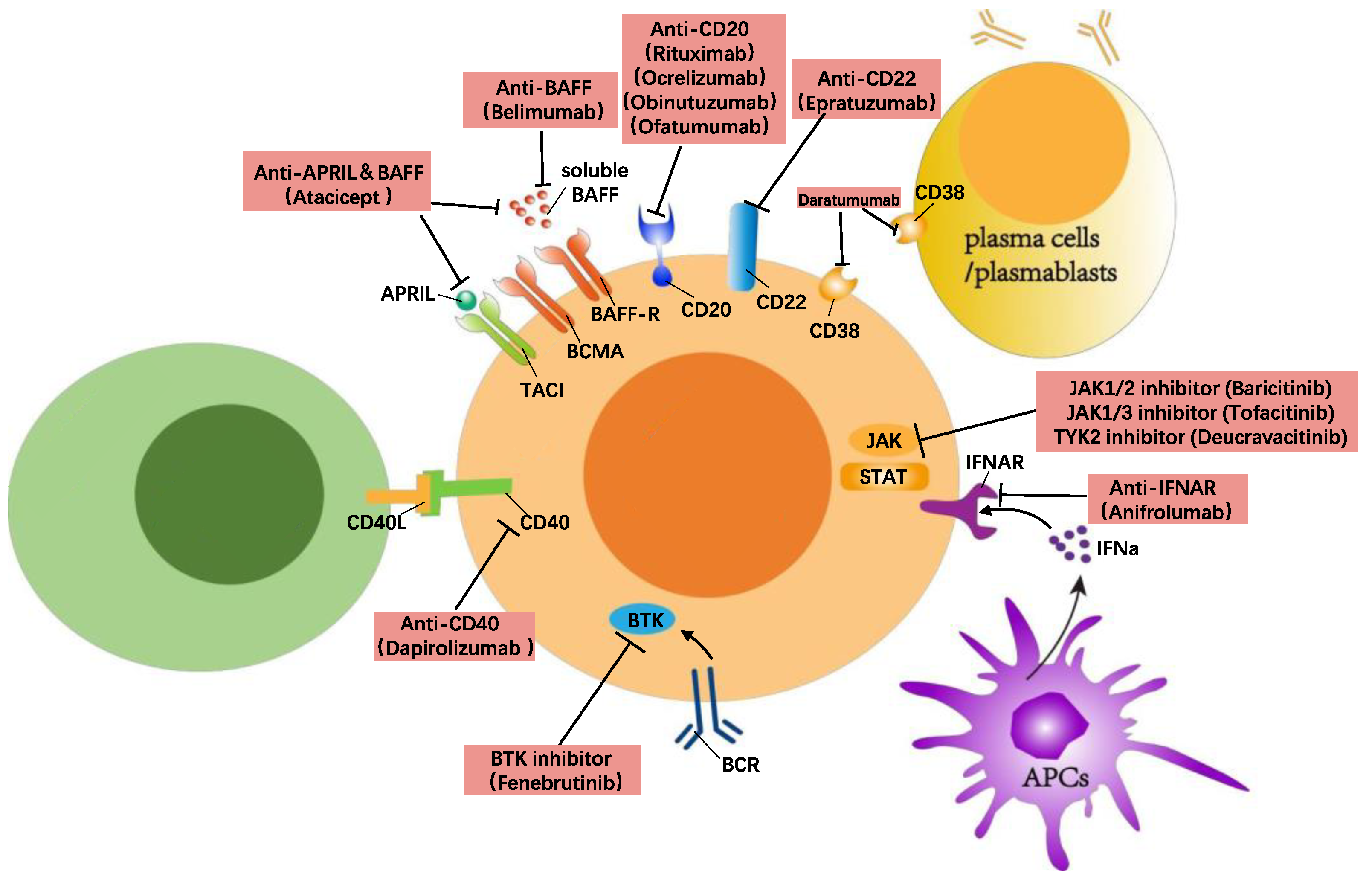
6. B Cell Directed Treatment in SLE
6.1. Anti-Inflammatory and Chemotherapy Treatments and Their Impact on B Cells
6.2. B Cell Suppressing/Depleting Therapies
6.2.1. Anti-BAFF/APRIL

{kind=link}
| Targets | Biologicals | Trial/Phase | Country | Number Enrolled | Primary Outcome | Result |
|---|---|---|---|---|---|---|
| BAFF | Belimumab | BLISS-76, Phase III (NCT00410384) | 136 centres primarily in 19 countries in North America and Europe. | 819 | SRI-4 at week 52. | Met the primary outcome [64]. |
| BAFF | Belimumab | BLISS-52, Phase III (NCT00424476) | Multicentres in Latin America, Asia-Pacific, and Eastern Europe. | 867 | SRI-4 at week 52. | Met the primary outcome [65]. |
| BAFF | Belimumab | BLISS-LN, Phase III (NCT01639339) | 107 centres in 21 countries. | 448 | Renal response at week 104. | Met the primary outcome [70]. |
| BAFF/APRIL | Atacicept | ADDRESS II, Phase IIb (NCT01972568) | Multicentres in Europe, Asia, North America, Central and South America. | 306 | SRI-4 at week 24. | Did not meet primary outcome but reduction in disease activity and severe flares was observed [71]. |
| BAFF/APRIL | Telitacicept | Phase III | Multicentres in China. | 249 | SRI-4 at week 48. | Met the primary outcome [72]. |
| CD20 | Rituximab | EXPLORER nonrenal trial, Phase II/III (NCT00137969) | 55 centres in North America. | 257 | Major/partial BILAG response at week 52. | Did not meet the primary outcome. Reduced risk and frequency of SLE flares was observed [74]. |
| CD20 | Rituximab | LUNAR renal trial, Phase III (NCT00282347) | Multicentres in US and Latin America. | 144 | Renal response using predefined parameters at week 52. | Did not meet the primary outcome. Responders had reduction in dsDNA and C3/C4 levels [75]. |
| CD20 | Ocrelizumab | Phase III (NCT00626197) | 123 centres in 23 countries in Latin America, Asia, Western Europe, Eastern Europe, US, Canada, and Africa. | 381 | Renal response rates at week 48. | Initial results suggested some efficacy in treating LN in seropositive patients. Phase III trial was terminated prematurely due to inefficacy [76]. |
| CD20 | Obinutuzumab | Phase II (NCT02550652) | 46 sites in 12 countries in USA, South America and Europe. | 125 | Complete Renal Response at Week 52. | Met the primary outcome [77]. |
| CD22 | Epratuzumab | ALLEVIATE-1 (SL0003; NCT00111306) and ALLEVIATE-2 (SL0004; NCT00383214), Phase III | ALLEVIATE-1: 16 centres in Europe, UK and USA. ALLEVIATE-2: 28 centres in Europe, UK and USA. | 36/54 | The revised primary endpoint was BILAG response with no treatment failure at week 12. | Discontinued prematurely due to interruption in supply of medication [78]. |
| CD22 | Epratuzumab | EMBODY 1 (NCT0 1262365) and EMBODY -2 (NCT01261793), Phase III | USA, Brazil, and Europe. | 793/791 | BICLA response rates at week 48. | Did not meet the primary outcome [79]. |
| CD38 | Daratumumab | Case serious | USA. | 2 | N/A | Induced substantial clinical responses in two active SLE patients [80]. |
6.2.2. Anti-CD20
6.2.3. Anti-CD22
6.2.4. Anti-CD38
6.3. B Cell Signalling Targeted Therapies
6.3.1. Anti-CD40/CD40L
6.3.2. Anti-IFNα
6.3.3. Anti-BTK
6.3.4. Anti-JAK
6.3.5. Anti-NIK
6.3.6. Anti-Pin-1
6.4. Cell Therapy (CAR T Cell Therapy)
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update omicronn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum Dis. 2021, 80, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Petkau, G.; Turner, M. Signalling circuits that direct early B-cell development. Biochem. J. 2019, 476, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Tull, T.J.; Pitcher, M.J.; Guesdon, W.; Siu, J.H.Y.; Lebrero-Fernandez, C.; Zhao, Y.; Petrov, N.; Heck, S.; Ellis, R.; Dhami, P.; et al. Human marginal zone B cell development from early T2 progenitors. J. Exp. Med. 2021, 218, e20202001. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Shinohara, H.; Baba, Y. B cell signaling and fate decision. Annu. Rev. Immunol. 2010, 28, 21–55. [Google Scholar] [CrossRef] [PubMed]
- Azevedo Portilho, N.; Scarfo, R.; Bertesago, E.; Ismailoglu, I.; Kyba, M.; Kobayashi, M.; Ditadi, A.; Yoshimoto, M. B1 lymphocytes develop independently of Notch signaling during mouse embryonic development. Development 2021, 148, dev199373. [Google Scholar] [CrossRef]
- Smith, F.L.; Baumgarth, N. B-1 cell responses to infections. Curr. Opin. Immunol. 2019, 57, 23–31. [Google Scholar] [CrossRef]
- Chadburn, A. The spleen: Anatomy and anatomical function. Semin Hematol. 2000, 37, 13–21. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef]
- Hendricks, J.; Bos, N.A.; Kroese, F.G.M. Heterogeneity of Memory Marginal Zone B Cells. Crit. Rev. Immunol. 2018, 38, 145–158. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Cyster, J.G. Transcriptional regulation of memory B cell differentiation. Nat. Rev. Immunol. 2021, 21, 209–220. [Google Scholar] [CrossRef]
- Fairfax, K.A.; Tsantikos, E.; Figgett, W.A.; Vincent, F.B.; Quah, P.S.; LePage, M.; Hibbs, M.L.; Mackay, F. BAFF-driven autoimmunity requires CD19 expression. J. Autoimmun. 2015, 62, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Enzler, T.; Bonizzi, G.; Silverman, G.J.; Otero, D.C.; Widhopf, G.F.; Anzelon-Mills, A.; Rickert, R.C.; Karin, M. Alternative and classical NF-kappa B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity 2006, 25, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Seagal, J.; Melamed, D. Role of receptor revision in forming a B cell repertoire. Clin. Immunol. 2002, 105, 1–8. [Google Scholar] [CrossRef]
- Meffre, E.; O’Connor, K.C. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev. 2019, 292, 90–101. [Google Scholar] [CrossRef]
- Burnett, D.L.; Reed, J.H.; Christ, D.; Goodnow, C.C. Clonal redemption and clonal anergy as mechanisms to balance B cell tolerance and immunity. Immunol. Rev. 2019, 292, 61–75. [Google Scholar] [CrossRef]
- El Shikh, M.E.; El Sayed, R.M.; Sukumar, S.; Szakal, A.K.; Tew, J.G. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol. 2010, 31, 205–211. [Google Scholar] [CrossRef]
- Manz, R.A.; Hauser, A.E.; Hiepe, F.; Radbruch, A. Maintenance of serum antibody levels. Annu. Rev. Immunol. 2005, 23, 367–386. [Google Scholar] [CrossRef]
- Degn, S.E.; van der Poel, C.E.; Firl, D.J.; Ayoglu, B.; Al Qureshah, F.A.; Bajic, G.; Mesin, L.; Reynaud, C.A.; Weill, J.C.; Utz, P.J.; et al. Clonal Evolution of Autoreactive Germinal Centers. Cell 2017, 170, 913–926.e19. [Google Scholar] [CrossRef]
- Bedard, M.; Shrestha, D.; Priestman, D.A.; Wang, Y.; Schneider, F.; Matute, J.D.; Iyer, S.S.; Gileadi, U.; Prota, G.; Kandasamy, M.; et al. Sterile activation of invariant natural killer T cells by ER-stressed antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23671–23681. [Google Scholar] [CrossRef]
- Mackay, F.; Browning, J.L. BAFF: A fundamental survival factor for B cells. Nat. Rev. Immunol. 2002, 2, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Mockel, T.; Basta, F.; Weinmann-Menke, J.; Schwarting, A. B cell activating factor (BAFF): Structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun. Rev. 2021, 20, 102736. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Zickert, A.; Sundelin, B.; Axelsson, M.; Gerhardsson, J.; Svenungsson, E.; Malmström, V.; Gunnarsson, I. Evaluation of B lymphocyte stimulator and a proliferation inducing ligand as candidate biomarkers in lupus nephritis based on clinical and histopathological outcome following induction therapy. Lupus Sci. Med. 2015, 2, e000061. [Google Scholar] [CrossRef]
- Petri, M.; Fu, W.; Ranger, A.; Allaire, N.; Cullen, P.; Magder, L.S.; Zhang, Y. Association between changes in gene signatures expression and disease activity among patients with systemic lupus erythematosus. BMC Med. Genom. 2019, 12, 4. [Google Scholar] [CrossRef]
- Hagglof, T.; Sedimbi, S.K.; Yates, J.L.; Parsa, R.; Salas, B.H.; Harris, R.A.; Leadbetter, E.A.; Karlsson, M.C. Neutrophils license iNKT cells to regulate self-reactive mouse B cell responses. Nat. Immunol. 2016, 17, 1407–1414. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Chang, P.P. Innate B cell helpers reveal novel types of antibody responses. Nat. Immunol. 2013, 14, 119–126. [Google Scholar] [CrossRef]
- Cornelis, R.; Chang, H.D.; Radbruch, A. Keeping up with the stress of antibody production: BAFF and APRIL maintain memory plasma cells. Curr. Opin. Immunol. 2021, 71, 97–102. [Google Scholar] [CrossRef]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K. Innate response activator B cells: Origins and functions. Int. Immunol. 2015, 27, 537–541. [Google Scholar] [CrossRef]
- Fillatreau, S.; Manfroi, B.; Dorner, T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Scholz, J.L.; Marshak-Rothstein, A.; Cancro, M.P. Molecular pattern recognition in peripheral B cell tolerance: Lessons from age-associated B cells. Curr. Opin. Immunol. 2019, 61, 33–38. [Google Scholar] [CrossRef]
- Rubtsova, K.; Rubtsov, A.V.; Thurman, J.M.; Mennona, J.M.; Kappler, J.W.; Marrack, P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J. Clin. Investig. 2017, 127, 1392–1404. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. Human regulatory B cells in health and disease: Therapeutic potential. J. Clin. Investig. 2017, 127, 772–779. [Google Scholar] [CrossRef]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19+CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef]
- Heinemann, K.; Wilde, B.; Hoerning, A.; Tebbe, B.; Kribben, A.; Witzke, O.; Dolff, S. Decreased IL-10+ regulatory B cells (Bregs) in lupus nephritis patients. Scand. J. Rheumatol. 2016, 45, 312–316. [Google Scholar] [CrossRef]
- Bosma, A.; Abdel-Gadir, A.; Isenberg, D.A.; Jury, E.C.; Mauri, C. Lipid-antigen presentation by CD1d+ B cells is essential for the maintenance of invariant natural killer T cells. Immunity 2012, 36, 477–490. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Medina, K.L.; Yokota, T.; Rossi, M.I.; Sakaguchi, N.; Comp, P.C.; Kincade, P.W. Early lymphoid progenitors in mouse and man are highly sensitive to glucocorticoids. Int. Immunol. 2005, 17, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, M.; Sakata, K.; Ishii, A.; Iwata, S.; Nakayamada, S.; Tanaka, Y. Hydroxychloroquine efficiently suppresses inflammatory responses of human class-switched memory B cells via Toll-like receptor 9 inhibition. Clin. Immunol. 2018, 195, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Hidalgo, M.; Jimeno, A. Mycophenolate mofetil: An update. Drugs Today 2009, 45, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schluter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 92. [Google Scholar] [CrossRef]
- Walsh, K.P.; Mills, K.H. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013, 34, 521–530. [Google Scholar] [CrossRef]
- Dongworth, D.W.; Klaus, G.G. Effects of cyclosporin A on the immune system of the mouse. I. Evidence for a direct selective effect of cyclosporin A on B cells responding to anti-immunoglobulin antibodies. Eur. J. Immunol. 1982, 12, 1018–1022. [Google Scholar] [CrossRef]
- Hilchey, S.P.; Palshikar, M.G.; Emo, J.A.; Li, D.; Garigen, J.; Wang, J.; Mendelson, E.S.; Cipolla, V.; Thakar, J.; Zand, M.S. Cyclosporine a directly affects human and mouse b cell migration in vitro by disrupting a hIF-1 αdependent, o2 sensing, molecular switch. BMC Immunol. 2020, 21, 13. [Google Scholar] [CrossRef]
- Bende, R.J.; Jochems, G.J.; Frame, T.H.; Klein, M.R.; van Eijk, R.V.; van Lier, R.A.; Zeijlemaker, W.P. Effects of IL-4, IL-5, and IL-6 on growth and immunoglobulin production of Epstein-Barr virus-infected human B cells. Cell Immunol. 1992, 143, 310–323. [Google Scholar] [CrossRef]
- Thomson, A.W. The effects of cyclosporin A on non-T cell components of the immune system. J. Autoimmun. 1992, 5 (Suppl. A), 167–176. [Google Scholar] [CrossRef] [PubMed]
- Hannah, J.; Casian, A.; D’Cruz, D. Tacrolimus use in lupus nephritis: A systematic review and meta-analysis. Autoimmun. Rev. 2016, 15, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.H.; Kim, K.W.; Yu, J.H.; Kim, B.M.; Choi, B.S.; Park, C.W.; Kim, Y.S.; Cho, M.L.; Yang, C.W. Decrease of immature B cell and interleukin-10 during early-post-transplant period in renal transplant recipients under tacrolimus based immunosuppression. Transpl. Immunol. 2014, 30, 159–167. [Google Scholar] [CrossRef]
- Kraaijeveld, R.; Li, Y.; Yan, L.; de Leur, K.; Dieterich, M.; Peeters, A.M.A.; Wang, L.; Shi, Y.; Baan, C.C. Inhibition of T Helper Cell Differentiation by Tacrolimus or Sirolimus Results in Reduced B-Cell Activation: Effects on T Follicular Helper Cells. Transplant. Proc. 2019, 51, 3463–3473. [Google Scholar] [CrossRef]
- Heidt, S.; Roelen, D.L.; Eijsink, C.; Eikmans, M.; van Kooten, C.; Claas, F.H.; Mulder, A. Calcineurin inhibitors affect B cell antibody responses indirectly by interfering with T cell help. Clin. Exp. Immunol. 2010, 159, 199–207. [Google Scholar] [CrossRef]
- Fan, J.; Luo, J.; Yan, C.; Hao, R.; Zhao, X.; Jia, R.; He, J.; Xu, D.; Miao, M.; Li, X. Methotrexate, combined with cyclophosphamide attenuates murine collagen induced arthritis by modulating the expression level of Breg and DCs. Mol. Immunol. 2017, 90, 106–117. [Google Scholar] [CrossRef]
- Treml, J.F.; Hao, Y.; Stadanlick, J.E.; Cancro, M.P. The BLyS family: Toward a molecular understanding of B cell homeostasis. Cell Biochem. Biophys. 2009, 53, 1–16. [Google Scholar] [CrossRef]
- Goenka, R.; Matthews, A.H.; Zhang, B.; O’Neill, P.J.; Scholz, J.L.; Migone, T.S.; Leonard, W.J.; Stohl, W.; Hershberg, U.; Cancro, M.P. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J. Exp. Med. 2014, 211, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.D.; Sarosi, I.; Xia, X.Z.; McCabe, S.; Miner, K.; Solovyev, I.; Hawkins, N.; Kelley, M.; Chang, D.; Van, G.; et al. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 3370–3375. [Google Scholar] [CrossRef]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef]
- Gross, J.A.; Johnston, J.; Mudri, S.; Enselman, R.; Dillon, S.R.; Madden, K.; Xu, W.; Parrish-Novak, J.; Foster, D.; Lofton-Day, C.; et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 2000, 404, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Zollars, E.; Bienkowska, J.; Czerkowicz, J.; Allaire, N.; Ranger, A.M.; Magder, L.; Zollars, E.; Bienkowska, J.; Czerkowicz, J.; Allaire, N.; et al. BAFF (B cell activating factor) transcript level in peripheral blood of patients with SLE is associated with same-day disease activity as well as global activity over the next year. Lupus Sci. Med. 2015, 2, e000063. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Stohl, W.; Ginzler, E.M.; Becker, M.; Mishra, N.; Chatham, W.; Merrill, J.T.; Weinstein, A.; McCune, W.J.; Zhong, J.; et al. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: A phase I trial in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2008, 10, R109. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzova, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Stohl, W.; Hiepe, F.; Latinis, K.M.; Thomas, M.; Scheinberg, M.A.; Clarke, A.; Aranow, C.; Wellborne, F.R.; Abud-Mendoza, C.; Hough, D.R.; et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2328–2337. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Petri, M.A.; Cervera, R.; Roth, D.A.; Ji, B.N.; Kleoudis, C.S.; Zhong, Z.J.; Freimuth, W. Belimumab in the treatment of systemic lupus erythematosus: High disease activity predictors of response. Ann. Rheum. Dis. 2012, 71, 1343–1349. [Google Scholar] [CrossRef]
- Anders, H.J.; Saxena, R.; Zhao, M.H.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus nephritis. Nat. Rev. Dis. Primers 2020, 6, 7. [Google Scholar] [CrossRef]
- Margiotta, D.P.E.; Basta, F.; Batani, V.; Afeltra, A. Belimumab and low-doses of mycophenolate mofetil as induction therapy of class IV lupus nephritis: Case series and literature review. BMC Nephrol. 2018, 19, 54. [Google Scholar] [CrossRef]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.K.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Wax, S.; Kao, A.; Fraser, P.A.; Chang, P.; Isenberg, D.; ADDRESS II Investigators. Efficacy and Safety of Atacicept in Patients with Systemic Lupus Erythematosus: Results of a Twenty-Four-Week, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Arm, Phase IIb Study. Arthritis Rheumatol. 2018, 70, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Telitacicept: First Approval. Drugs 2021, 81, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Xue, R.; Zhou, X.; Shen, P.; Wang, S.; Yang, Y. Telitacicept as a BLyS/APRIL dual inhibitor for autoimmune disease. Immunopharmacol. Immunotoxicol. 2021, 43, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Mysler, E.F.; Spindler, A.J.; Guzman, R.; Bijl, M.; Jayne, D.; Furie, R.A.; Houssiau, F.A.; Drappa, J.; Close, D.; Maciuca, R.; et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: Results from a randomized, double-blind, phase III study. Arthritis Rheum. 2013, 65, 2368–2379. [Google Scholar] [CrossRef]
- Furie, R.A.; Aroca, G.; Cascino, M.D.; Garg, J.P.; Rovin, B.H.; Alvarez, A.; Fragoso-Loyo, H.; Zuta-Santillan, E.; Schindler, T.; Brunetta, P.; et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: A randomised, double-blind, placebo-controlled trial. Ann. Rheum Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef]
- Wallace, D.J.; Gordon, C.; Strand, V.; Hobbs, K.; Petri, M.; Kalunian, K.; Houssiau, F.; Tak, P.P.; Isenberg, D.A.; Kelley, L.; et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic lupus erythematosus: Results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology 2013, 52, 1313–1322. [Google Scholar] [CrossRef]
- Clowse, M.E.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczynski, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results from Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar] [CrossRef]
- Ostendorf, L.; Burns, M.; Durek, P.; Heinz, G.A.; Heinrich, F.; Garantziotis, P.; Enghard, P.; Richter, U.; Biesen, R.; Schneider, U.; et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 383, 1149–1155. [Google Scholar] [CrossRef]
- Smith, K.G.; Jones, R.B.; Burns, S.M.; Jayne, D.R. Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: Remission, relapse, and re-treatment. Arthritis Rheum. 2006, 54, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Ginzler, E.M.; Merrill, J.T.; Furie, R.A.; Stohl, W.; Chatham, W.W.; Weinstein, A.; McKay, J.D.; McCune, W.J.; Petri, M.; et al. Safety and Efficacy of Belimumab Plus Standard Therapy for Up to Thirteen Years in Patients with Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Masoud, S.; McAdoo, S.P.; Bedi, R.; Cairns, T.D.; Lightstone, L. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology 2018, 57, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, G.; Isenberg, D.A.; Edwards, J.C.; Leandro, M.J.; Migone, T.S.; Teodorescu, M.; Stohl, W. B cell depletion therapy in systemic lupus erythematosus: Relationships among serum B lymphocyte stimulator levels, autoantibody profile and clinical response. Ann. Rheum. Dis. 2008, 67, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Wing, C. The BAFFling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef]
- Vallerskog, T.; Heimburger, M.; Gunnarsson, I.; Zhou, W.; Wahren-Herlenius, M.; Trollmo, C.; Malmström, V. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R167. [Google Scholar] [CrossRef]
- Kraaij, T.; Kamerling, S.W.A.; de Rooij, E.N.M.; van Daele, P.L.A.; Bredewold, O.W.; Bakker, J.A.; Bajema, I.M.; Scherer, H.U.; Toes, R.E.M.; Huizinga, T.J.W.; et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J. Autoimmun. 2018, 91, 45–54. [Google Scholar] [CrossRef]
- SM, Y.A.-F.; Harris, K.M.; Byron, M.; Ding, L.; Kanaparthi, S.; Barry, W.T.; Gao, W.; Ryker, K.; Tosta, P.; Askanase, A.D.; et al. Phase II Randomized Trial of Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis. Arthritis Rheumatol. 2021, 73, 121–131. [Google Scholar]
- Dorner, T.; Kaufmann, J.; Wegener, W.A.; Teoh, N.; Goldenberg, D.M.; Burmester, G.R. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res. Ther. 2006, 8, R74. [Google Scholar] [CrossRef]
- Pavon, E.J.; Zumaquero, E.; Rosal-Vela, A.; Khoo, K.M.; Cerezo-Wallis, D.; Garcia-Rodriguez, S.; Carrascal, M.; Abian, J.; Graeff, R.; Callejas-Rubio, J.L.; et al. Increased CD38 expression in T cells and circulating anti-CD38 IgG autoantibodies differentially correlate with distinct cytokine profiles and disease activity in systemic lupus erythematosus patients. Cytokine 2013, 62, 232–243. [Google Scholar] [CrossRef]
- Plesner, T.; Krejcik, J. Daratumumab for the Treatment of Multiple Myeloma. Front Immunol. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed]
- Benfaremo, D.; Gabrielli, A. Is There a Future for Anti-CD38 Antibody Therapy in Systemic Autoimmune Diseases? Cells 2019, 9, 77. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kuhl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Walhelm, T.; Gunnarsson, I.; Heijke, R.; Leonard, D.; Trysberg, E.; Eriksson, P.; Sjöwall, C. Clinical Experience of Proteasome Inhibitor Bortezomib Regarding Efficacy and Safety in Severe Systemic Lupus Erythematosus: A Nationwide Study. Front. Immunol. 2021, 12, 756941. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Perper, S.J.; Westmoreland, S.V.; Karman, J.; Twomey, R.; Seagal, J.; Wang, R.; McRae, B.L.; Clarke, S.H. Treatment with a CD40 Antagonist Antibody Reverses Severe Proteinuria and Loss of Saliva Production and Restores Glomerular Morphology in Murine Systemic Lupus Erythematosus. J. Immunol. 2019, 203, 58–75. [Google Scholar] [CrossRef]
- Furie, R.A.; Bruce, I.N.; Dorner, T.; Leon, M.G.; Leszczynski, P.; Urowitz, M.; Haier, B.; Jimenez, T.; Brittain, C.; Liu, J.; et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 2021, 60, 5397–5407. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 2021, 12, 3391. [Google Scholar] [CrossRef]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dörner, T.; Cardiel, M.H.; Bruce, I.N.; Gomez, E.; et al. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.; Pike, M.; Merrill, J.T.; van Vollenhoven, R.; Werth, V.P.; Hobar, C.; Delev, N.; Shah, V.; Sharkey, B.; Wegman, T.; et al. Deucravacitinib, a Tyrosine Kinase 2 Inhibitor, in Systemic Lupus Erythematosus: A Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2023, 75, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor Fenebrutinib (GDC-0853) in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef]
- Klarquist, J.; Cantrell, R.; Lehn, M.A.; Lampe, K.; Hennies, C.M.; Hoebe, K.; Janssen, E.M. Type I IFN Drives Experimental Systemic Lupus Erythematosus by Distinct Mechanisms in CD4 T Cells and B Cells. Immunohorizons 2020, 4, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Gillanders, R.N.; Arzhakova, O.V.; Hempel, A.; Dolgova, A.; Kerry, J.P.; Yarysheva, L.M.; Bakeev, N.F.; Volynskii, A.L.; Papkovsky, D.B. Phosphorescent oxygen sensors based on nanostructured polyolefin substrates. Anal. Chem. 2010, 82, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; CD1067 study investigators. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- Chatham, W.W.; Furie, R.; Saxena, A.; Brohawn, P.; Schwetje, E.; Abreu, G.; Tummala, R. Long-Term Safety and Efficacy of Anifrolumab in Adults with Systemic Lupus Erythematosus: Results of a Phase II Open-Label Extension Study. Arthritis Rheumatol. 2021, 73, 816–825. [Google Scholar] [CrossRef]
- Chia, Y.L.; Santiago, L.; Wang, B.; Kuruvilla, D.; Wang, S.; Tummala, R.; Roskos, L. Exposure-response analysis for selection of optimal dosage regimen of anifrolumab in patients with systemic lupus erythematosus. Rheumatology 2021, 60, 5854–5862. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves AstraZeneca’s anifrolumab for lupus. Nat. Rev. Drug. Discov. 2021, 20, 658. [Google Scholar] [CrossRef]
- Mina-Osorio, P.; LaStant, J.; Keirstead, N.; Whittard, T.; Ayala, J.; Stefanova, S.; Garrido, R.; Dimaano, N.; Hilton, H.; Giron, M.; et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013, 65, 2380–2391. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Efraim, M.; Mayer, J.; Trneny, M.; McDonald, V.; Bird, R.; Regenbogen, T.; Garg, M.; Kaplan, Z.; Tzvetkov, N.; et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N. Engl. J. Med. 2022, 386, 1421–1431. [Google Scholar] [CrossRef]
- Maurer, M.; Berger, W.; Gimenez-Arnau, A.; Hayama, K.; Jain, V.; Reich, A.; Haemmerle, S.; Lheritier, K.; Walsh, P.; Xia, S.; et al. Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria. J. Allergy Clin. Immunol. 2022, 150, 1498–1506.e2. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.R.; Schlissel, M.S. Partial restoration of B cell development in Jak-3-/- mice achieved by co-expression of IgH and Eμ-myc transgenes. Int. Immunol. 2002, 14, 893–904. [Google Scholar] [CrossRef]
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Abdalrahman, Z.; Sciumè, G.; Tsai, W.L.; Trier, A.M.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017, 69, 148–160. [Google Scholar] [CrossRef]
- Rizzi, M.; Lorenzetti, R.; Fischer, K.; Staniek, J.; Janowska, I.; Troilo, A.; Strohmeier, V.; Erlacher, M.; Kunze, M.; Bannert, B.; et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J. Autoimmun. 2017, 77, 55–66. [Google Scholar] [CrossRef]
- Yan, Q.; Chen, W.; Song, H.; Long, X.; Zhang, Z.; Tang, X.; Chen, H.; Lin, H.; Sun, L. Tofacitinib Ameliorates Lupus through Suppression of T Cell Activation Mediated by TGF-Beta Type I Receptor. Front. Immunol. 2021, 12, 675542. [Google Scholar] [CrossRef]
- Kubo, S.; Yamaoka, K.; Kondo, M.; Yamagata, K.; Zhao, J.; Iwata, S.; Tanaka, Y. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann. Rheum. Dis. 2014, 73, 2192–2198. [Google Scholar] [CrossRef]
- Nikolopoulos, D.; Parodis, I. Janus kinase inhibitors in systemic lupus erythematosus: Implications for tyrosine kinase 2 inhibition. Front. Med. 2023, 10, 1217147. [Google Scholar] [CrossRef]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug. Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Brightbill, H.D.; Suto, E.; Blaquiere, N.; Ramamoorthi, N.; Sujatha-Bhaskar, S.; Gogol, E.B.; Castanedo, G.M.; Jackson, B.T.; Kwon, Y.C.; Haller, S.; et al. NF-kappaB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun. 2018, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yoshida, N.; Finn, G.; Kozono, S.; Nechama, M.; Kyttaris, V.C.; Zhen Zhou, X.; Tsokos, G.C.; Ping Lu, K. Pin1-Targeted Therapy for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Garimella, M.G.; He, C.; Chen, G.; Li, Q.Z.; Huang, X.; Karlsson, M.C.I. The B cell response to both protein and nucleic acid antigens displayed on apoptotic cells are dependent on endosomal pattern recognition receptors. J. Autoimmun. 2021, 117, 102582. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Kronke, G.; Volkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef]
- Mackensen, A.; Muller, F.; Mougiakakos, D.; Boltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]
- Kamburova, E.G.; Koenen, H.J.; Borgman, K.J.; ten Berge, I.J.; Joosten, I.; Hilbrands, L.B. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am. J. Transplant. 2013, 13, 1503–1511. [Google Scholar] [CrossRef]
- Thurlings, R.M.; Vos, K.; Wijbrandts, C.A.; Zwinderman, A.H.; Gerlag, D.M.; Tak, P.P. Synovial tissue response to rituximab: Mechanism of action and identification of biomarkers of response. Ann. Rheum. Dis. 2008, 67, 917–925. [Google Scholar] [CrossRef]
| Targets | Biologicals | Trial/Phase | Country | Number Enrolled | Primary Outcome | Result |
|---|---|---|---|---|---|---|
| CD40/CD40L | Dapirolizumab | Phase IIb (NCT02804763) | Europe, Latin America, and North America. | 182 | Dose–response based on BICLA responder rates at week 24 | Did not meet the primary outcome but reduction in disease activity and severe flares was observed [98]. |
| IFNAR | Anifrolumab | TULIP–2, Phase III (NCT02446899) | 119 sites in 16 countries. | 362 | BICLA response at week 52 | Met the primary outcome [99]. |
| JAK1/3 | Tofacitinib | Phase I (NCT02535689) | USA. | 30 | Safety of tofacitinib in SLE subjects (time frame: 5 years) | A dose of tofacitinib (5 mg twice daily) is safe and well tolerated in SLE patients [100]. |
| JAK1/2 | Baricitinib | BRAVE I, Phase II (NCT02708095) | 78 centres in 11 countries in Asia, Europe, North America, and South America. | 314 | SLEDAI-2K arthritis or rash resolution at week 24 | Met the primary outcome [101]. |
| TYK2 | Deucravacitinib | Phase II (NCT03252587) | 162 sites in 17 countries in Asia, Europe, North America, and South America. | 363 | SRI-4 response at week 32 | Met the primary outcome [102]. |
| BTK | Fenebrutinib | Phase II (NCT02908100) | 44 sites in 12 countries mainly in Latin America, the US, and Western Europe. | 260 | SRI-4 response at week 48 | Did not meet the primary outcome, but significantly reduced levels of CD19-positive B cells, including plasmablasts [103]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parodis, I.; Long, X.; Karlsson, M.C.I.; Huang, X. B Cell Tolerance and Targeted Therapies in SLE. J. Clin. Med. 2023, 12, 6268. https://doi.org/10.3390/jcm12196268
Parodis I, Long X, Karlsson MCI, Huang X. B Cell Tolerance and Targeted Therapies in SLE. Journal of Clinical Medicine. 2023; 12(19):6268. https://doi.org/10.3390/jcm12196268
Chicago/Turabian StyleParodis, Ioannis, Xuan Long, Mikael C. I. Karlsson, and Xin Huang. 2023. "B Cell Tolerance and Targeted Therapies in SLE" Journal of Clinical Medicine 12, no. 19: 6268. https://doi.org/10.3390/jcm12196268
APA StyleParodis, I., Long, X., Karlsson, M. C. I., & Huang, X. (2023). B Cell Tolerance and Targeted Therapies in SLE. Journal of Clinical Medicine, 12(19), 6268. https://doi.org/10.3390/jcm12196268